Department of Computer Science and Engineering, University of Connecticut, 371 Fairfield Way, U-2155, Storrs, CT 06269, USA.
BMC Bioinformatics. 2010 Nov 15;11:560. doi: 10.1186/1471-2105-11-560.
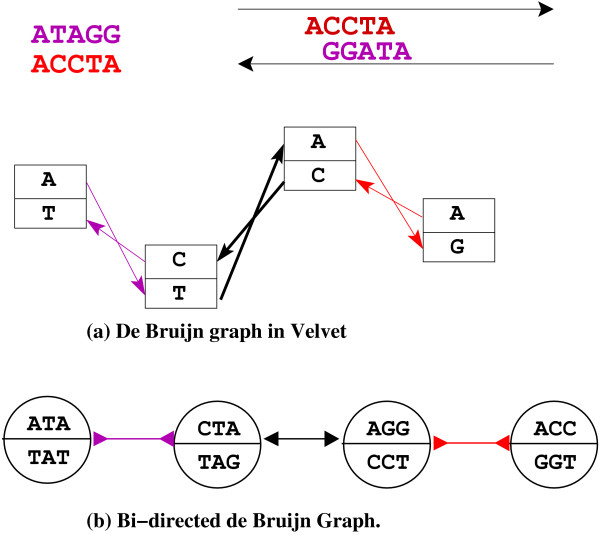
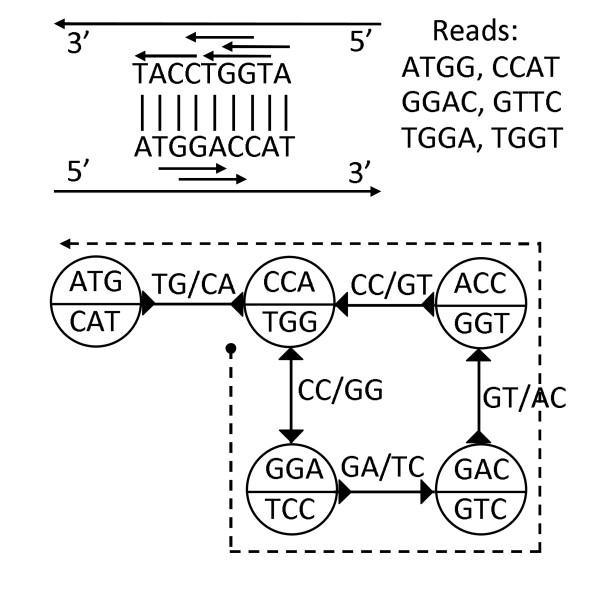
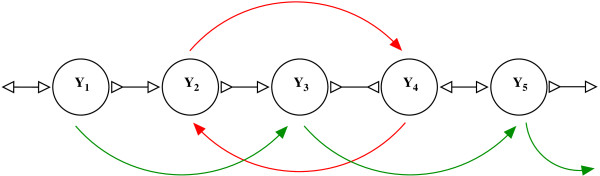
Assembling genomic sequences from a set of overlapping reads is one of the most fundamental problems in computational biology. Algorithms addressing the assembly problem fall into two broad categories - based on the data structures which they employ. The first class uses an overlap/string graph and the second type uses a de Bruijn graph. However with the recent advances in short read sequencing technology, de Bruijn graph based algorithms seem to play a vital role in practice. Efficient algorithms for building these massive de Bruijn graphs are very essential in large sequencing projects based on short reads. In an earlier work, an O(n/p) time parallel algorithm has been given for this problem. Here n is the size of the input and p is the number of processors. This algorithm enumerates all possible bi-directed edges which can overlap with a node and ends up generating Θ(nΣ) messages (Σ being the size of the alphabet).
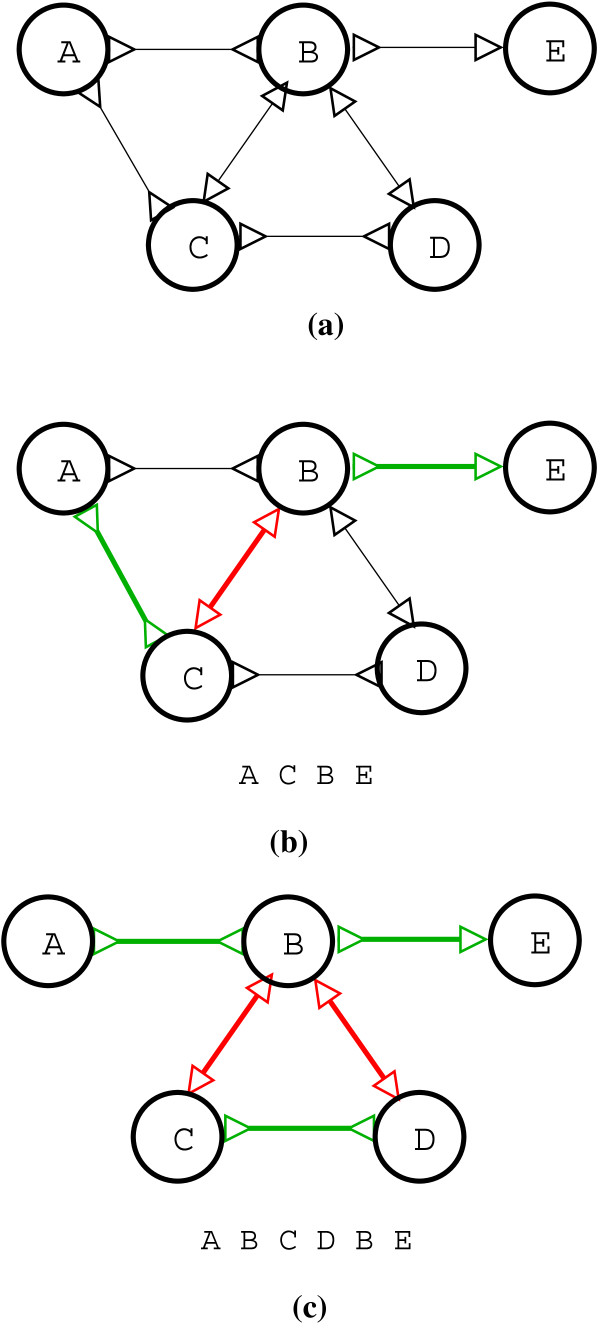
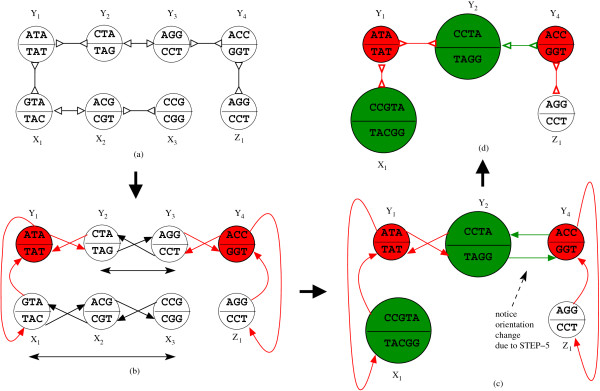
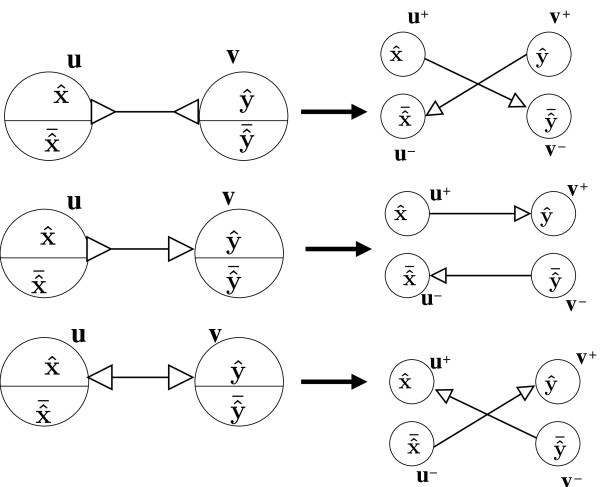
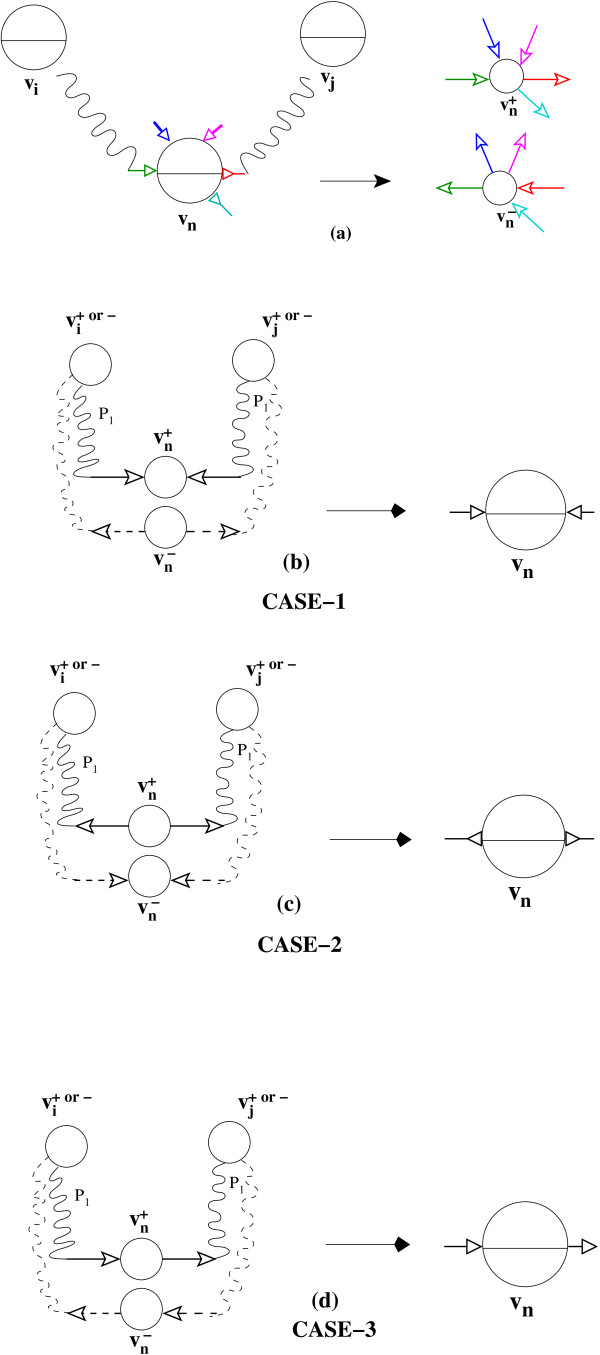
In this paper we present a Θ(n/p) time parallel algorithm with a communication complexity that is equal to that of parallel sorting and is not sensitive to Σ. The generality of our algorithm makes it very easy to extend it even to the out-of-core model and in this case it has an optimal I/O complexity of Θ(nlog(n/B)Blog(M/B)) (M being the main memory size and B being the size of the disk block). We demonstrate the scalability of our parallel algorithm on a SGI/Altix computer. A comparison of our algorithm with the previous approaches reveals that our algorithm is faster--both asymptotically and practically. We demonstrate the scalability of our sequential out-of-core algorithm by comparing it with the algorithm used by VELVET to build the bi-directed de Bruijn graph. Our experiments reveal that our algorithm can build the graph with a constant amount of memory, which clearly outperforms VELVET. We also provide efficient algorithms for the bi-directed chain compaction problem.
The bi-directed de Bruijn graph is a fundamental data structure for any sequence assembly program based on Eulerian approach. Our algorithms for constructing Bi-directed de Bruijn graphs are efficient in parallel and out of core settings. These algorithms can be used in building large scale bi-directed de Bruijn graphs. Furthermore, our algorithms do not employ any all-to-all communications in a parallel setting and perform better than the prior algorithms. Finally our out-of-core algorithm is extremely memory efficient and can replace the existing graph construction algorithm in VELVET.
将一组重叠读取的基因组序列组装在一起是计算生物学中最基本的问题之一。解决组装问题的算法分为两类 - 基于它们使用的数据结构。第一类使用重叠/字符串图,第二类使用 de Bruijn 图。然而,随着短读测序技术的最新进展,基于 de Bruijn 图的算法在实践中似乎起着至关重要的作用。在基于短读的大型测序项目中,构建这些大规模 de Bruijn 图的高效算法是非常必要的。在早期的工作中,已经给出了一种用于此问题的 O(n/p)时间并行算法。这里 n 是输入的大小,p 是处理器的数量。该算法枚举可以与节点重叠的所有可能的双向边,并最终生成 Θ(nΣ)条消息(Σ 是字母表的大小)。
在本文中,我们提出了一种具有与并行排序相同通信复杂度的 Θ(n/p)时间并行算法,并且对 Σ 不敏感。我们算法的通用性使得它非常容易扩展到核外模型,在这种情况下,它具有最优的 I/O 复杂度为 Θ(nlog(n/B)Blog(M/B))(M 是主内存大小,B 是磁盘块的大小)。我们在 SGI/Altix 计算机上展示了我们的并行算法的可扩展性。将我们的算法与以前的方法进行比较表明,我们的算法无论是在理论上还是在实践中都更快。我们通过将其与 VELVET 用于构建双向 de Bruijn 图的算法进行比较,展示了我们的顺序核外算法的可扩展性。我们的实验表明,我们的算法可以使用恒定数量的内存构建图形,这明显优于 VELVET。我们还提供了用于双向链紧缩问题的有效算法。
双向 de Bruijn 图是基于 Eulerian 方法的任何序列组装程序的基本数据结构。我们用于构建双向 de Bruijn 图的算法在并行和核外环境中都非常高效。这些算法可用于构建大规模双向 de Bruijn 图。此外,我们的算法在并行设置中不使用任何全对全通信,并且比以前的算法表现更好。最后,我们的核外算法具有极高的内存效率,可以替代 VELVET 中现有的图构建算法。