Limasset Antoine, Cazaux Bastien, Rivals Eric, Peterlongo Pierre
IRISA Inria Rennes Bretagne Atlantique, GenScale team, Campus de Beaulieu, Rennes, 35042, France.
L.I.R.M.M., UMR 5506, Université de Montpellier et CNRS, 860 rue de St Priest, Montpellier Cedex 5, F-34392, France.
BMC Bioinformatics. 2016 Jun 16;17(1):237. doi: 10.1186/s12859-016-1103-9.
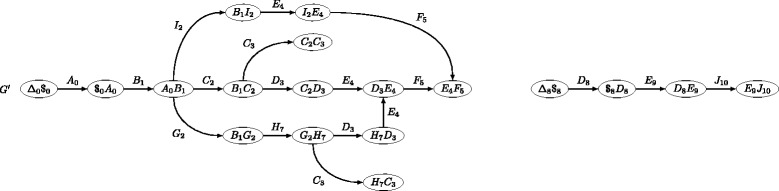
Next Generation Sequencing (NGS) has dramatically enhanced our ability to sequence genomes, but not to assemble them. In practice, many published genome sequences remain in the state of a large set of contigs. Each contig describes the sequence found along some path of the assembly graph, however, the set of contigs does not record all the sequence information contained in that graph. Although many subsequent analyses can be performed with the set of contigs, one may ask whether mapping reads on the contigs is as informative as mapping them on the paths of the assembly graph. Currently, one lacks practical tools to perform mapping on such graphs.
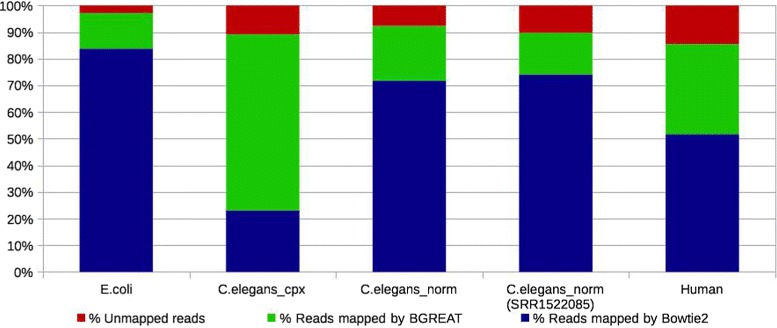
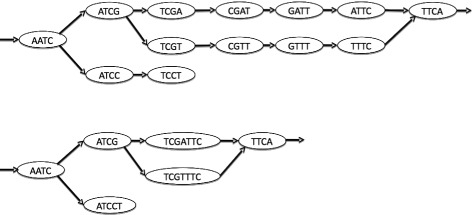
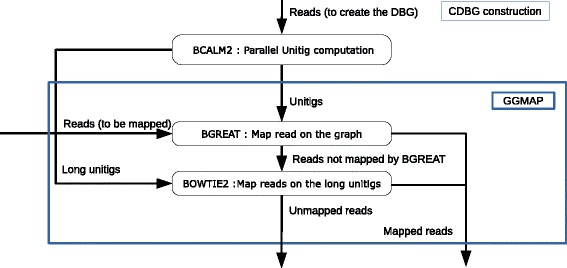
Here, we propose a formal definition of mapping on a de Bruijn graph, analyse the problem complexity which turns out to be NP-complete, and provide a practical solution. We propose a pipeline called GGMAP (Greedy Graph MAPping). Its novelty is a procedure to map reads on branching paths of the graph, for which we designed a heuristic algorithm called BGREAT (de Bruijn Graph REAd mapping Tool). For the sake of efficiency, BGREAT rewrites a read sequence as a succession of unitigs sequences. GGMAP can map millions of reads per CPU hour on a de Bruijn graph built from a large set of human genomic reads. Surprisingly, results show that up to 22 % more reads can be mapped on the graph but not on the contig set.
Although mapping reads on a de Bruijn graph is complex task, our proposal offers a practical solution combining efficiency with an improved mapping capacity compared to assembly-based mapping even for complex eukaryotic data.
新一代测序(NGS)极大地提升了我们对基因组进行测序的能力,但在基因组组装方面却并非如此。实际上,许多已发表的基因组序列仍处于大量重叠群的状态。每个重叠群描述了沿着组装图的某些路径所发现的序列,然而,重叠群集合并未记录该图中包含的所有序列信息。尽管可以使用重叠群集合进行许多后续分析,但有人可能会问,将 reads 映射到重叠群上是否与将它们映射到组装图的路径上一样具有信息量。目前,缺乏在这样的图上进行映射的实用工具。
在此,我们提出了在德布鲁因图上进行映射的形式化定义,分析了结果证明是 NP 完全问题的复杂度,并提供了一个实际解决方案。我们提出了一个名为 GGMAP(贪婪图映射)的流程。它的新颖之处在于一种将 reads 映射到图的分支路径上的程序,为此我们设计了一种名为 BGREAT(德布鲁因图 reads 映射工具)的启发式算法。为了提高效率,BGREAT 将一个 reads 序列重写为一系列单重叠群序列。GGMAP 每 CPU 小时可以在由大量人类基因组 reads 构建的德布鲁因图上映射数百万个 reads。令人惊讶的是,结果表明,在图上可以映射的 reads 比在重叠群集合上多 22%。
尽管将 reads 映射到德布鲁因图上是一项复杂的任务,但我们的提议提供了一个实际解决方案,与基于组装的映射相比,即使对于复杂的真核生物数据,也能将效率与改进的映射能力相结合。