Abba M C, Lacunza E, Butti M, Aldaz C M
Centro de Investigaciones Inmunológicas Básicas y Aplicadas (CINIBA), Facultad de Ciencias Médicas, Universidad Nacional de La Plata, Argentina.
Biomark Insights. 2010 Oct 27;5:103-18. doi: 10.4137/BMI.S5740.
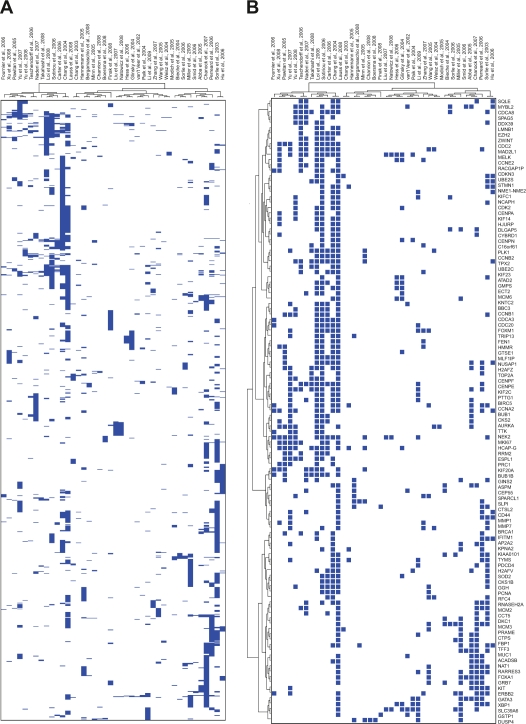
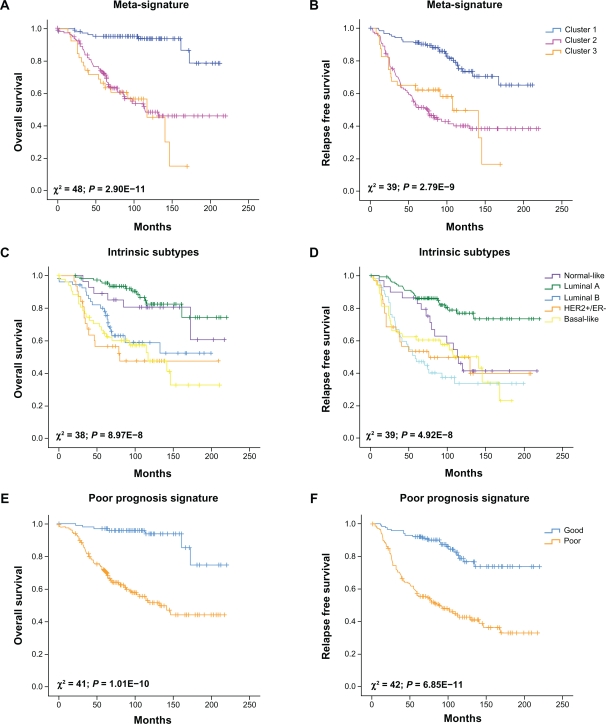
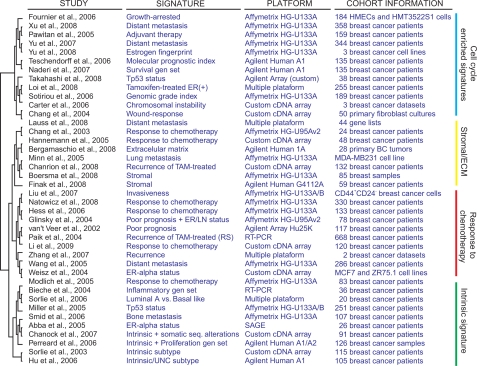
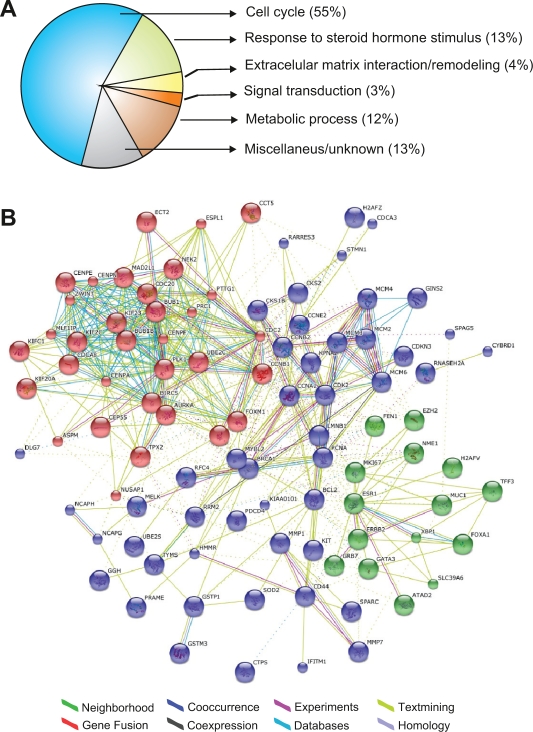
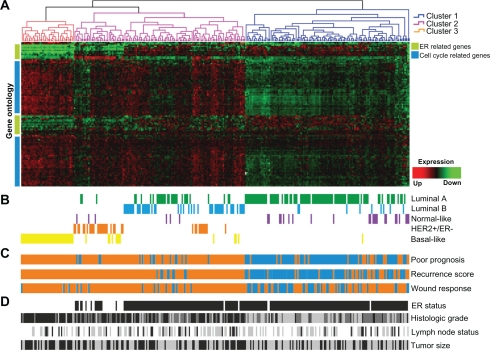
In this review we provide a systematic analysis of transcriptomic signatures derived from 42 breast cancer gene expression studies, in an effort to identify the most relevant breast cancer biomarkers using a meta-analysis method. Meta-data revealed a set of 117 genes that were the most commonly affected ranging from 12% to 36% of overlap among breast cancer gene expression studies. Data mining analysis of transcripts and protein-protein interactions of these commonly modulated genes indicate three functional modules significantly affected among signatures, one module related with the response to steroid hormone stimulus, and two modules related to the cell cycle. Analysis of a publicly available gene expression data showed that the obtained meta-signature is capable of predicting overall survival (P < 0.0001) and relapse-free survival (P < 0.0001) in patients with early-stage breast carcinomas. In addition, the identified meta-signature improves breast cancer patient stratification independently of traditional prognostic factors in a multivariate Cox proportional-hazards analysis.
在本综述中,我们对来自42项乳腺癌基因表达研究的转录组特征进行了系统分析,旨在使用荟萃分析方法识别最相关的乳腺癌生物标志物。元数据揭示了一组117个基因,这些基因在乳腺癌基因表达研究中的重叠率最常受到影响,范围从12%到36%。对这些常见调控基因的转录本和蛋白质-蛋白质相互作用进行数据挖掘分析表明,在特征中三个功能模块受到显著影响,一个模块与对类固醇激素刺激的反应相关,另外两个模块与细胞周期相关。对公开可用的基因表达数据的分析表明,所获得的元特征能够预测早期乳腺癌患者的总生存期(P < 0.0001)和无复发生存期(P < 0.0001)。此外,在多变量Cox比例风险分析中,所识别的元特征独立于传统预后因素改善了乳腺癌患者的分层。