Department of Health Studies, University of Chicago, Chicago, Illinois, United States of America.
PLoS Genet. 2010 Nov 11;6(11):e1001202. doi: 10.1371/journal.pgen.1001202.
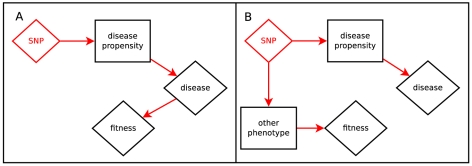
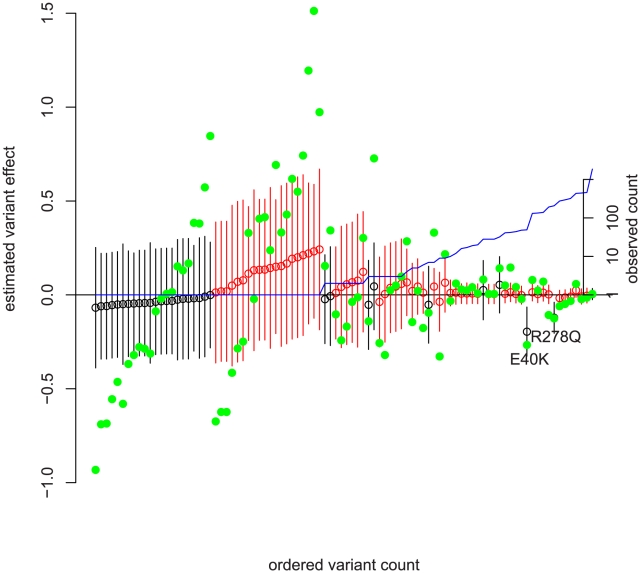
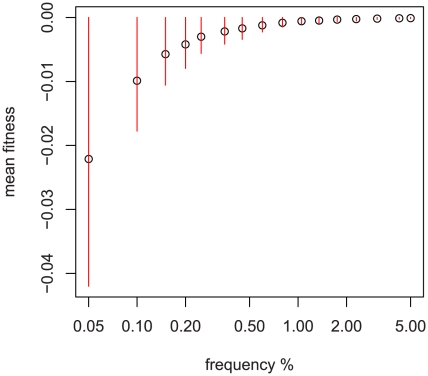
Sequencing technologies are becoming cheap enough to apply to large numbers of study participants and promise to provide new insights into human phenotypes by bringing to light rare and previously unknown genetic variants. We develop a new framework for the analysis of sequence data that incorporates all of the major features of previously proposed approaches, including those focused on allele counts and allele burden, but is both more general and more powerful. We harness population genetic theory to provide prior information on effect sizes and to create a pooling strategy for information from rare variants. Our method, EMMPAT (Evolutionary Mixed Model for Pooled Association Testing), generates a single test per gene (substantially reducing multiple testing concerns), facilitates graphical summaries, and improves the interpretation of results by allowing calculation of attributable variance. Simulations show that, relative to previously used approaches, our method increases the power to detect genes that affect phenotype when natural selection has kept alleles with large effect sizes rare. We demonstrate our approach on a population-based re-sequencing study of association between serum triglycerides and variation in ANGPTL4.
测序技术变得足够廉价,可以应用于大量的研究参与者,并有望通过揭示罕见和以前未知的遗传变异,为人类表型提供新的见解。我们开发了一种新的序列数据分析框架,它结合了以前提出的方法的所有主要特征,包括专注于等位基因计数和等位基因负担的方法,但更具普遍性和更强的功能。我们利用群体遗传理论来提供关于效应大小的先验信息,并为来自罕见变异的信息创建一个汇集策略。我们的方法,即 EMMPAT(用于合并关联测试的进化混合模型),为每个基因生成一个单一的测试(大大减少了多重测试的问题),便于图形汇总,并通过允许计算可归因方差来改善结果的解释。模拟表明,与以前使用的方法相比,当自然选择使具有大效应大小的等位基因变得罕见时,我们的方法增加了检测影响表型的基因的能力。我们在一项基于人群的 ANGPTL4 血清甘油三酯与变异之间关联的重测序研究中展示了我们的方法。