Theoretical Biology and Biophysics and Center for Nonlinear Studies, Los Alamos National Laboratory, Los Alamos, New Mexico, USA.
PLoS Genet. 2009 Oct;5(10):e1000695. doi: 10.1371/journal.pgen.1000695. Epub 2009 Oct 23.
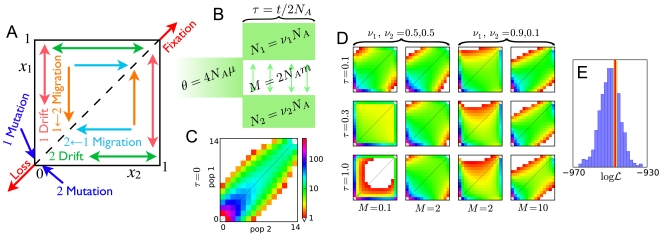
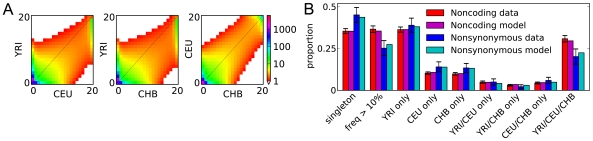
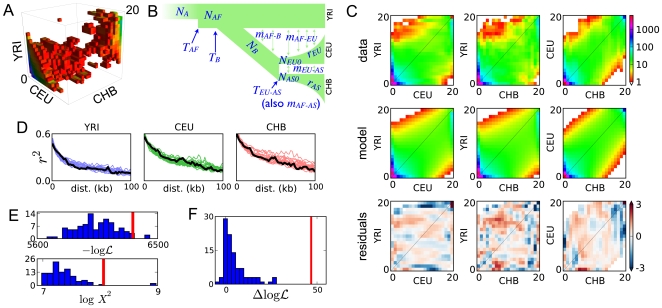
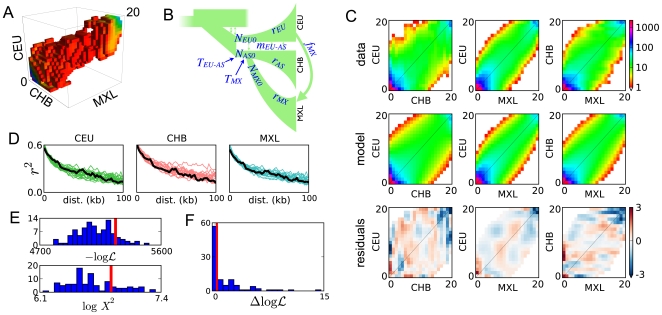
Demographic models built from genetic data play important roles in illuminating prehistorical events and serving as null models in genome scans for selection. We introduce an inference method based on the joint frequency spectrum of genetic variants within and between populations. For candidate models we numerically compute the expected spectrum using a diffusion approximation to the one-locus, two-allele Wright-Fisher process, involving up to three simultaneous populations. Our approach is a composite likelihood scheme, since linkage between neutral loci alters the variance but not the expectation of the frequency spectrum. We thus use bootstraps incorporating linkage to estimate uncertainties for parameters and significance values for hypothesis tests. Our method can also incorporate selection on single sites, predicting the joint distribution of selected alleles among populations experiencing a bevy of evolutionary forces, including expansions, contractions, migrations, and admixture. We model human expansion out of Africa and the settlement of the New World, using 5 Mb of noncoding DNA resequenced in 68 individuals from 4 populations (YRI, CHB, CEU, and MXL) by the Environmental Genome Project. We infer divergence between West African and Eurasian populations 140 thousand years ago (95% confidence interval: 40-270 kya). This is earlier than other genetic studies, in part because we incorporate migration. We estimate the European (CEU) and East Asian (CHB) divergence time to be 23 kya (95% c.i.: 17-43 kya), long after archeological evidence places modern humans in Europe. Finally, we estimate divergence between East Asians (CHB) and Mexican-Americans (MXL) of 22 kya (95% c.i.: 16.3-26.9 kya), and our analysis yields no evidence for subsequent migration. Furthermore, combining our demographic model with a previously estimated distribution of selective effects among newly arising amino acid mutations accurately predicts the frequency spectrum of nonsynonymous variants across three continental populations (YRI, CHB, CEU).
基于遗传数据构建的人口统计学模型在阐明史前事件和作为基因组选择扫描的零模型方面发挥着重要作用。我们引入了一种基于群体内和群体间遗传变异联合频率谱的推断方法。对于候选模型,我们使用涉及三个同时群体的单基因座、双等位基因 Wright-Fisher 过程的扩散逼近来数值计算预期谱。我们的方法是一种复合似然方案,因为中性基因座之间的连锁改变了频率谱的方差,但没有改变其期望。因此,我们使用包含连锁的自举来估计参数的不确定性和假设检验的显著性值。我们的方法还可以纳入对单一位点的选择,预测经历多种进化力量(包括扩张、收缩、迁移和混合)的群体中选定等位基因的联合分布。我们模拟了人类从非洲的扩张和新世界的定居,使用环境基因组计划中 68 个人的 4 个群体(YRI、CHB、CEU 和 MXL)中重新测序的 5 Mb 非编码 DNA。我们推断出西非和欧亚人群之间的分歧发生在 14 万年前(95%置信区间:40-270 kya)。这比其他遗传研究更早,部分原因是我们纳入了迁移。我们估计欧洲(CEU)和东亚(CHB)的分歧时间为 23 kya(95%置信区间:17-43 kya),远在考古证据将现代人置于欧洲之后。最后,我们估计东亚人(CHB)和墨西哥裔美国人(MXL)之间的分歧时间为 22 kya(95%置信区间:16.3-26.9 kya),我们的分析没有发现随后迁移的证据。此外,将我们的人口统计学模型与先前估计的新出现的氨基酸突变中选择效应的分布相结合,准确地预测了三个大陆群体(YRI、CHB、CEU)中非同义变体的频率谱。