Center for Genomics and Systems Biology, Department of Biology, New York University, New York, NY 10003, USA.
BMC Evol Biol. 2010 Nov 18;10:357. doi: 10.1186/1471-2148-10-357.
Gene duplication can lead to genetic redundancy, which masks the function of mutated genes in genetic analyses. Methods to increase sensitivity in identifying genetic redundancy can improve the efficiency of reverse genetics and lend insights into the evolutionary outcomes of gene duplication. Machine learning techniques are well suited to classifying gene family members into redundant and non-redundant gene pairs in model species where sufficient genetic and genomic data is available, such as Arabidopsis thaliana, the test case used here.
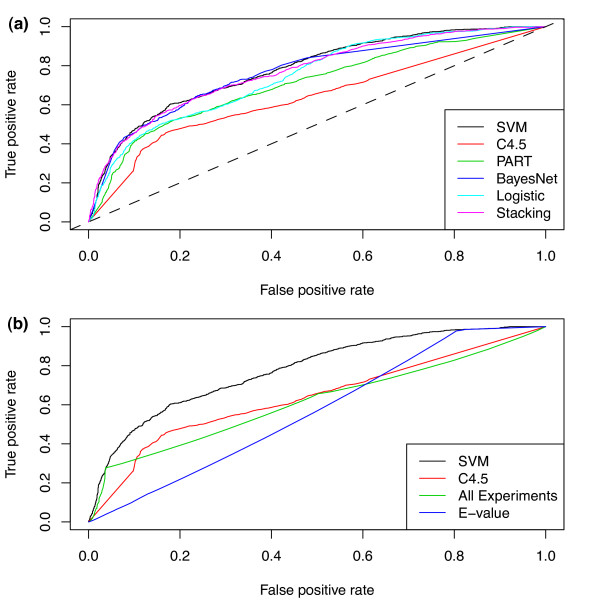
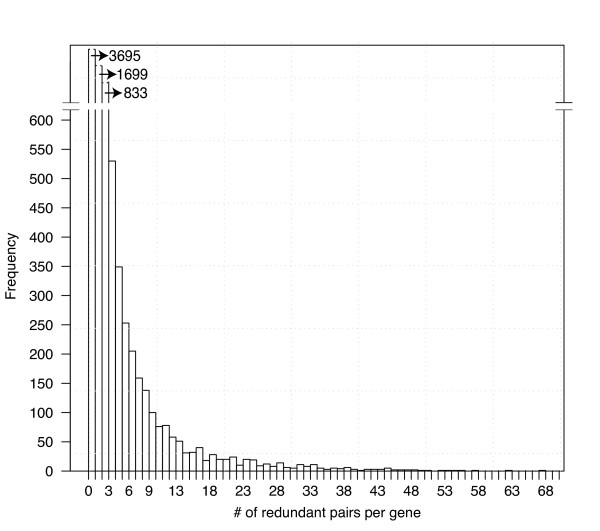
Machine learning techniques that combine multiple attributes led to a dramatic improvement in predicting genetic redundancy over single trait classifiers alone, such as BLAST E-values or expression correlation. In withholding analysis, one of the methods used here, Support Vector Machines, was two-fold more precise than single attribute classifiers, reaching a level where the majority of redundant calls were correctly labeled. Using this higher confidence in identifying redundancy, machine learning predicts that about half of all genes in Arabidopsis showed the signature of predicted redundancy with at least one but typically less than three other family members. Interestingly, a large proportion of predicted redundant gene pairs were relatively old duplications (e.g., Ks > 1), suggesting that redundancy is stable over long evolutionary periods.
Machine learning predicts that most genes will have a functionally redundant paralog but will exhibit redundancy with relatively few genes within a family. The predictions and gene pair attributes for Arabidopsis provide a new resource for research in genetics and genome evolution. These techniques can now be applied to other organisms.
基因复制可能导致遗传冗余,从而掩盖遗传分析中突变基因的功能。提高识别遗传冗余敏感性的方法可以提高反向遗传学的效率,并深入了解基因复制的进化结果。机器学习技术非常适合在具有足够遗传和基因组数据的模式物种(如这里使用的拟南芥)中将基因家族成员分类为冗余和非冗余基因对。
将多个属性结合起来的机器学习技术,与仅使用单个特征分类器(如 BLAST E 值或表达相关性)相比,极大地提高了预测遗传冗余的能力。在保留分析中,这里使用的一种方法——支持向量机,比单属性分类器精确两倍,达到了大多数冗余调用都被正确标记的水平。通过这种更高的冗余识别置信度,机器学习预测约有一半的拟南芥基因与至少一个但通常少于三个其他家族成员具有预测冗余的特征。有趣的是,很大一部分预测的冗余基因对是相对较老的复制(例如,Ks > 1),这表明冗余在较长的进化时期是稳定的。
机器学习预测大多数基因将具有功能冗余的同源基因,但在家族内与相对较少的基因表现出冗余。拟南芥的预测和基因对属性为遗传学和基因组进化研究提供了新的资源。这些技术现在可以应用于其他生物体。