Center of Excellence in Environmental Toxicology, Department of Pharmacology, University of Pennsylvania School of Medicine, 130C John Morgan Bldg, 3620 Hamilton Walk, Philadelphia, PA 19104-6084, United States.
J Steroid Biochem Mol Biol. 2011 May;125(1-2):95-104. doi: 10.1016/j.jsbmb.2010.11.004. Epub 2010 Nov 16.
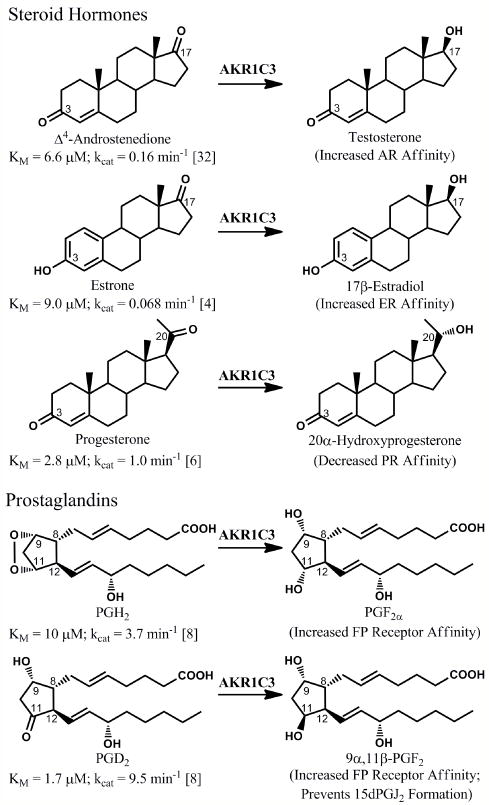
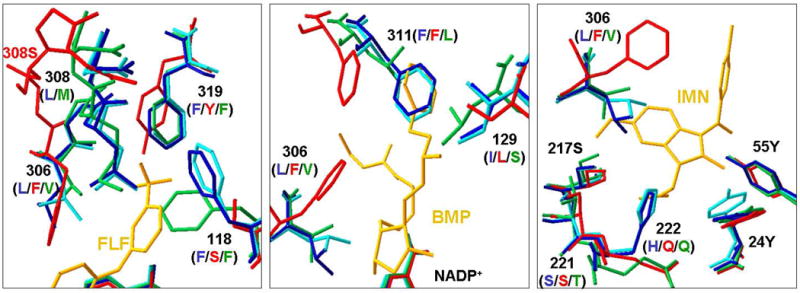
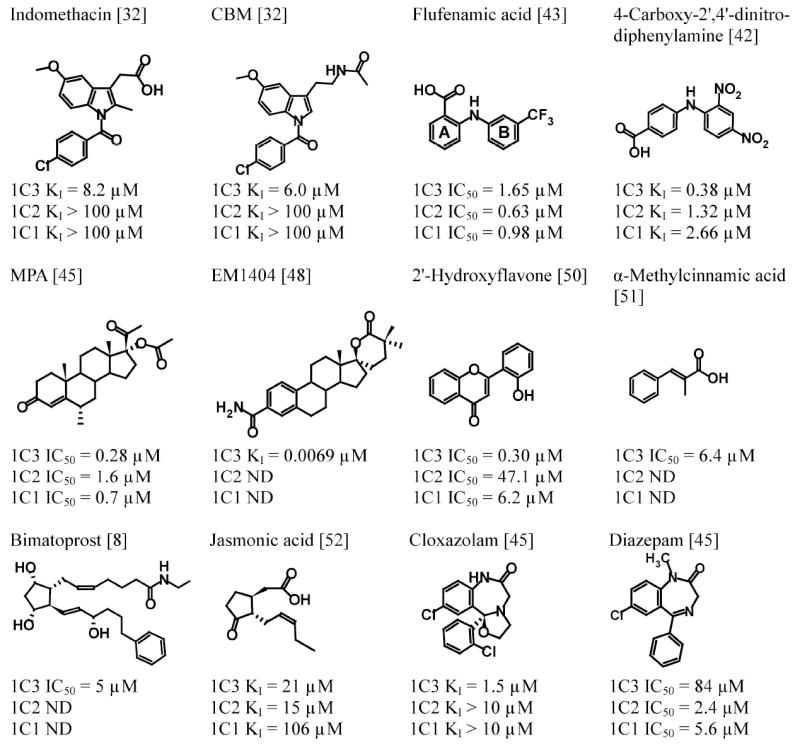
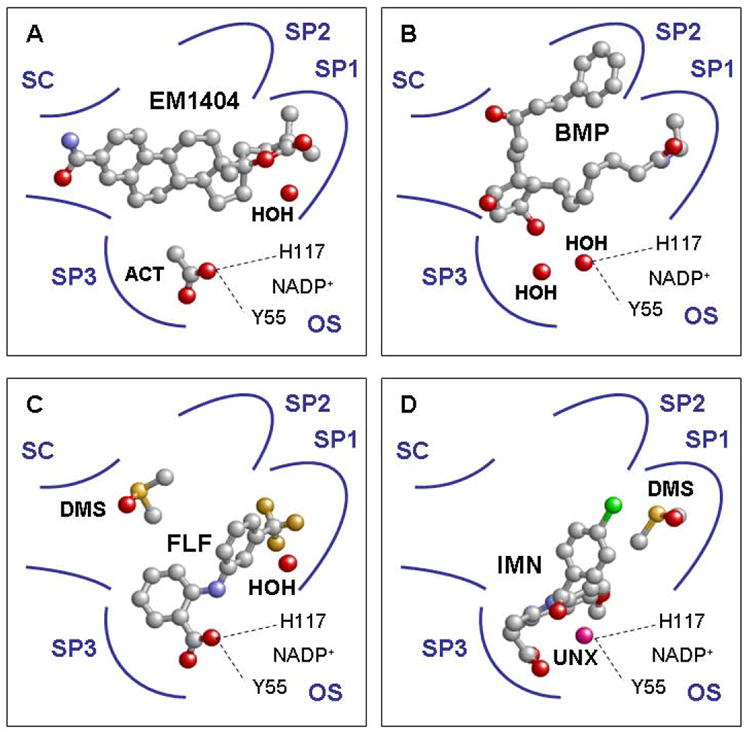
There is considerable interest in the development of an inhibitor of aldo-keto reductase (AKR) 1C3 (type 5 17β-hydroxysteroid dehydrogenase and prostaglandin F synthase) as a potential therapeutic for both hormone-dependent and hormone-independent cancers. AKR1C3 catalyzes the reduction of 4-androstene-3,17-dione to testosterone and estrone to 17β-estradiol in target tissues, which will promote the proliferation of hormone dependent prostate and breast cancers, respectively. AKR1C3 also catalyzes the reduction of prostaglandin (PG) H(2) to PGF(2α) and PGD(2) to 9α,11β-PGF(2), which will limit the formation of anti-proliferative prostaglandins, including 15-deoxy-Δ(12,14)-PGJ(2), and contribute to proliferative signaling. AKR1C3 is overexpressed in a wide variety of cancers, including breast and prostate cancer. An inhibitor of AKR1C3 should not inhibit the closely related isoforms AKR1C1 and AKR1C2, as they are involved in other key steroid hormone biotransformations in target tissues. Several structural leads have been explored as inhibitors of AKR1C3, including non-steroidal anti-inflammatory drugs, steroid hormone analogues, flavonoids, cyclopentanes, and benzodiazepines. Inspection of the available crystal structures of AKR1C3 with multiple ligands bound, along with the crystal structures of the other AKR1C isoforms, provides a structural basis for the rational design of isoform specific inhibitors of AKR1C3. We find that there are subpockets involved in ligand binding that are considerably different in AKR1C3 relative to the closely related AKR1C1 or AKR1C2 isoforms. These pockets can be used to further improve the binding affinity and selectivity of the currently available AKR1C3 inhibitors. Article from the special issue on Targeted Inhibitors.
人们对醛酮还原酶 1C3(类型 5 17β-羟甾脱氢酶和前列腺素 F 合酶)抑制剂的开发很感兴趣,因为它可能成为治疗激素依赖性和非依赖性癌症的潜在药物。AKR1C3 在靶组织中将 4-雄烯-3,17-二酮还原为睾酮,将雌酮还原为 17β-雌二醇,这将分别促进激素依赖性前列腺癌和乳腺癌的增殖。AKR1C3 还催化前列腺素(PG)H(2)还原为 PGF(2α)和 PGD(2)为 9α,11β-PGF(2),这将限制抗增殖前列腺素的形成,包括 15-脱氧-Δ(12,14)-PGJ(2),并促进增殖信号。AKR1C3 在多种癌症中过度表达,包括乳腺癌和前列腺癌。AKR1C3 的抑制剂不应抑制密切相关的同工酶 AKR1C1 和 AKR1C2,因为它们参与了靶组织中其他关键的甾体激素生物转化。已经探索了几种结构先导物作为 AKR1C3 的抑制剂,包括非甾体抗炎药、甾体激素类似物、类黄酮、环戊烷和苯并二氮䓬。结合多个配体结合的 AKR1C3 以及其他 AKR1C 同工酶的晶体结构的检查,为 AKR1C3 的同工酶特异性抑制剂的合理设计提供了结构基础。我们发现,与密切相关的 AKR1C1 或 AKR1C2 同工酶相比,AKR1C3 中的配体结合涉及到相当不同的亚口袋。这些口袋可用于进一步提高现有 AKR1C3 抑制剂的结合亲和力和选择性。本文选自靶向抑制剂特刊。