Auckland Cancer Society Research Centre, University of Auckland, Auckland, New Zealand.
PLoS One. 2012;7(8):e43965. doi: 10.1371/journal.pone.0043965. Epub 2012 Aug 28.
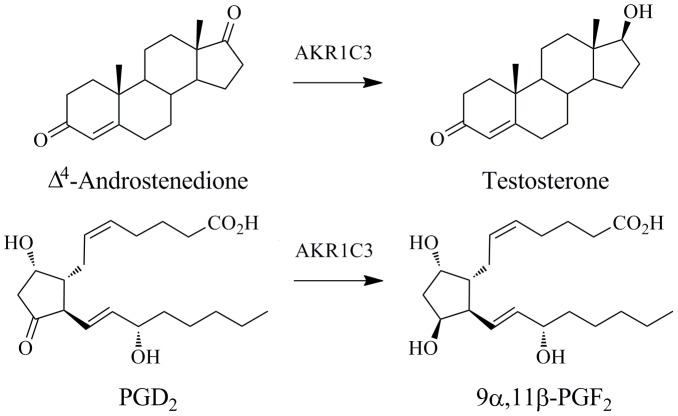
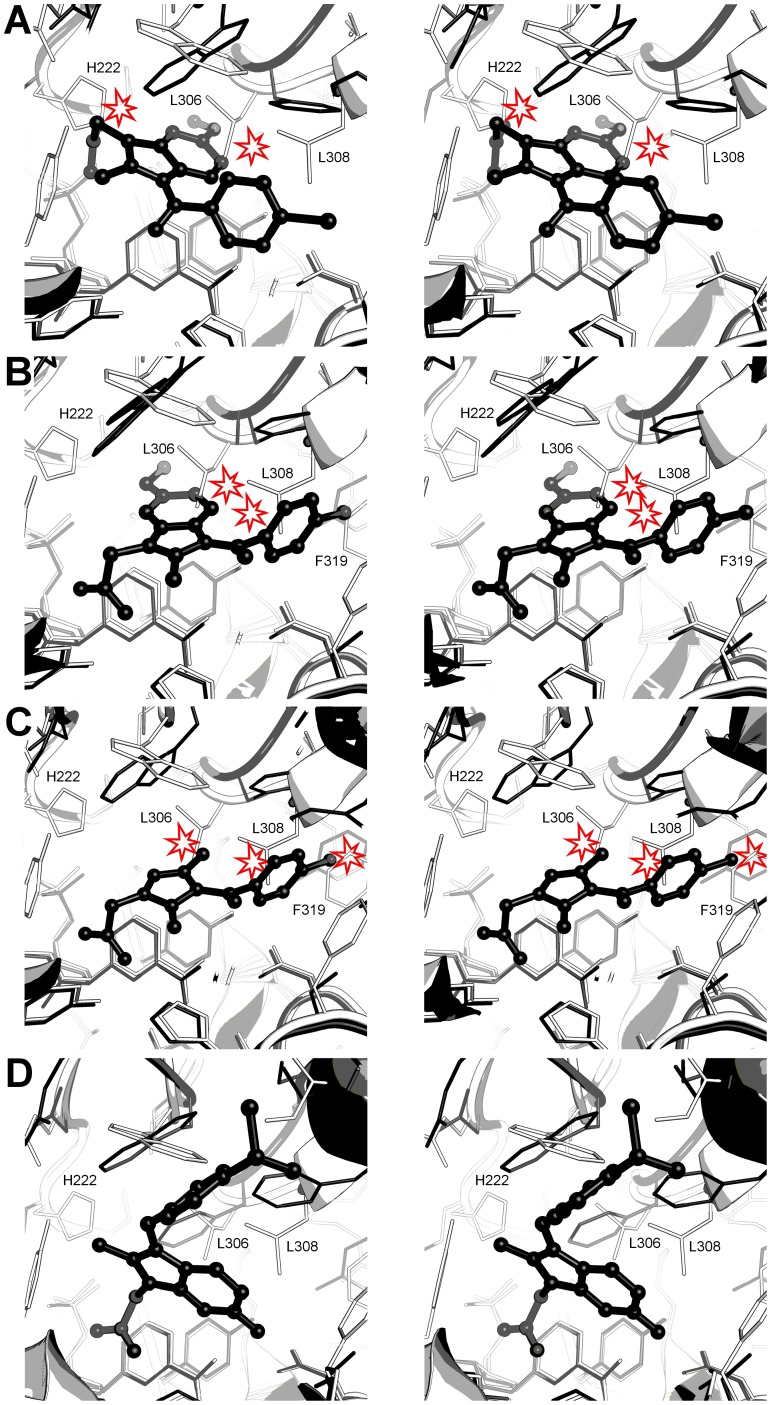
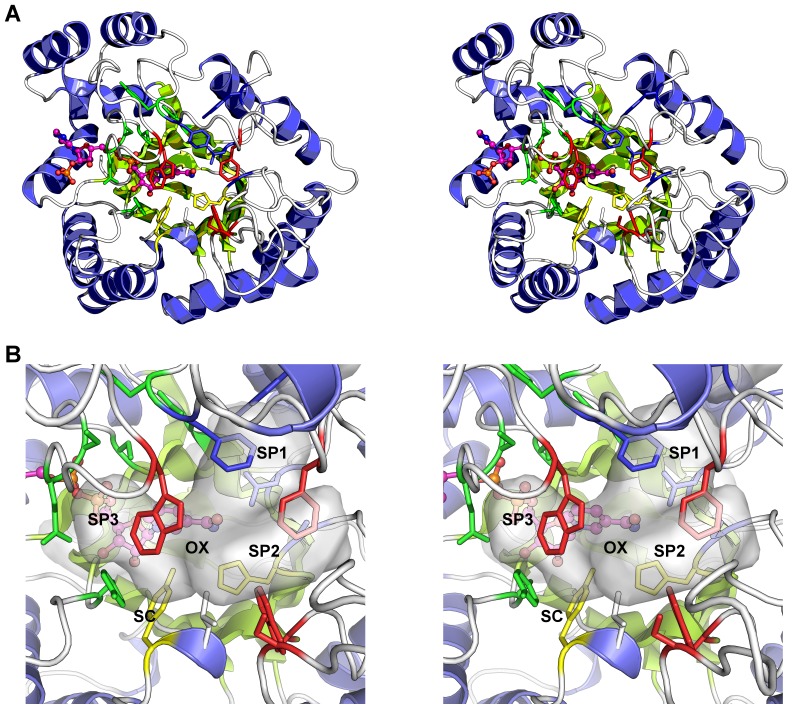
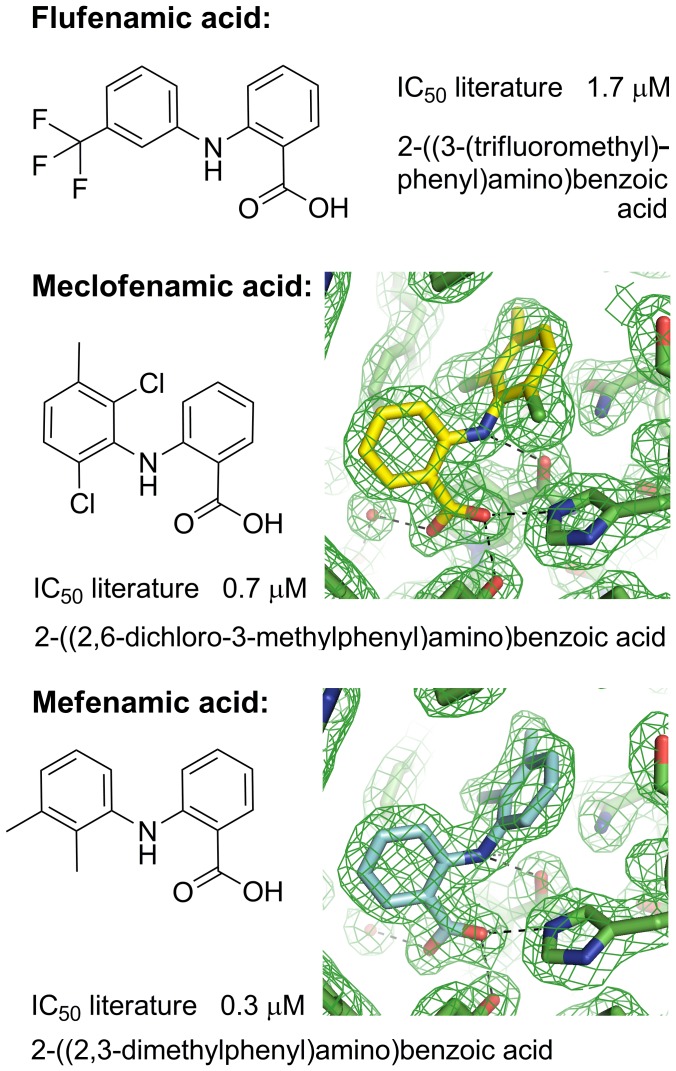
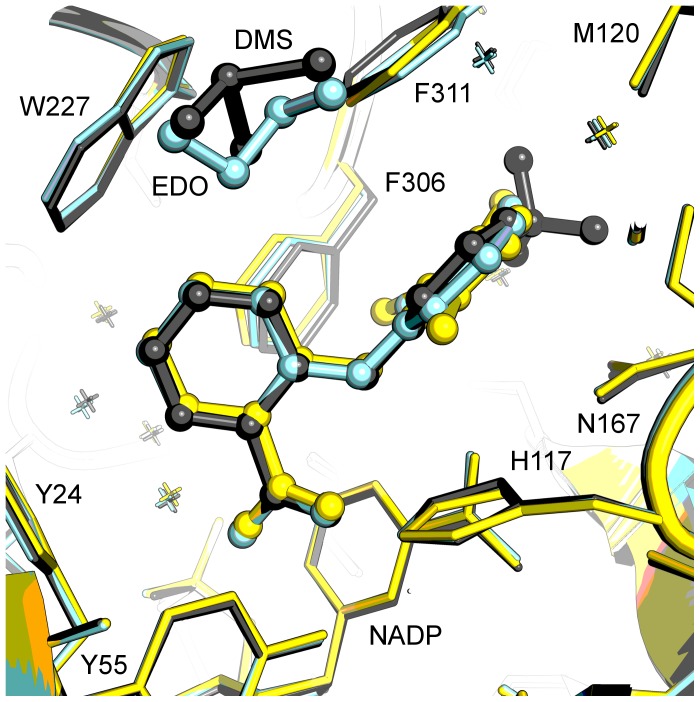
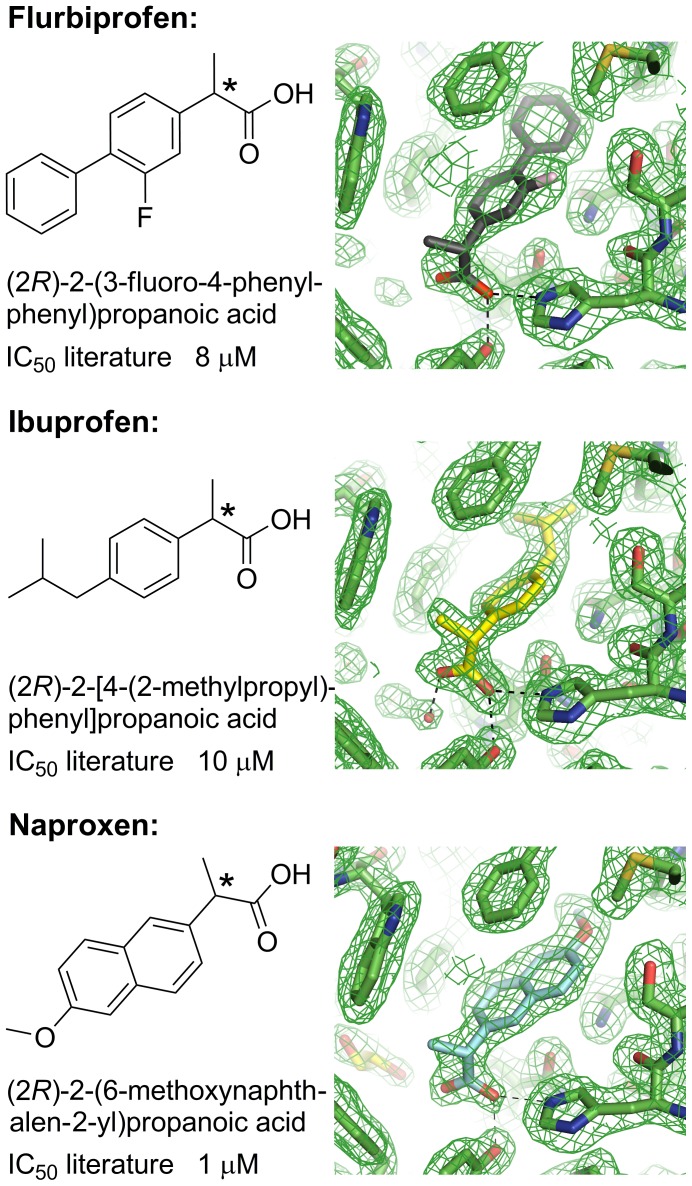
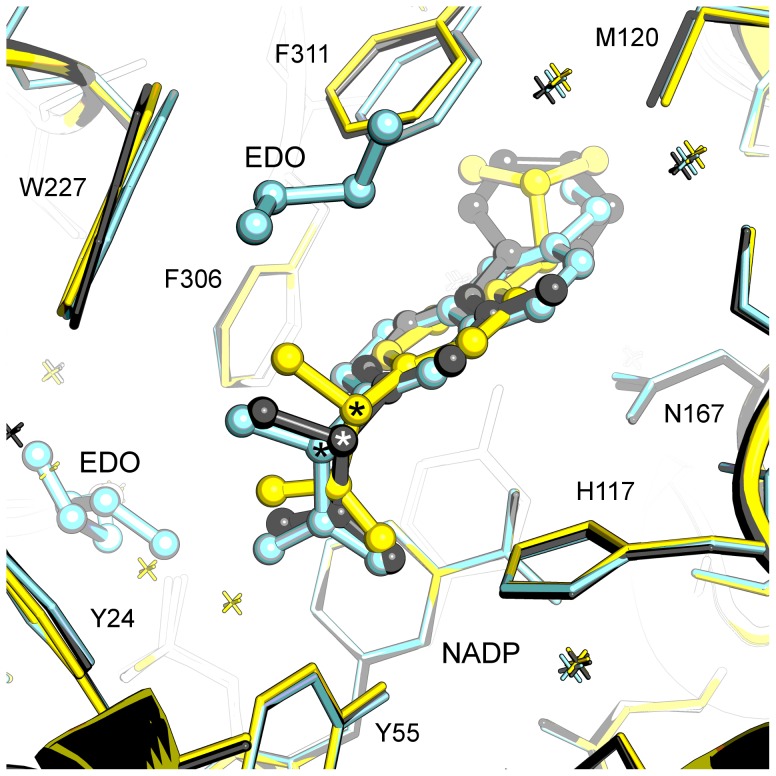
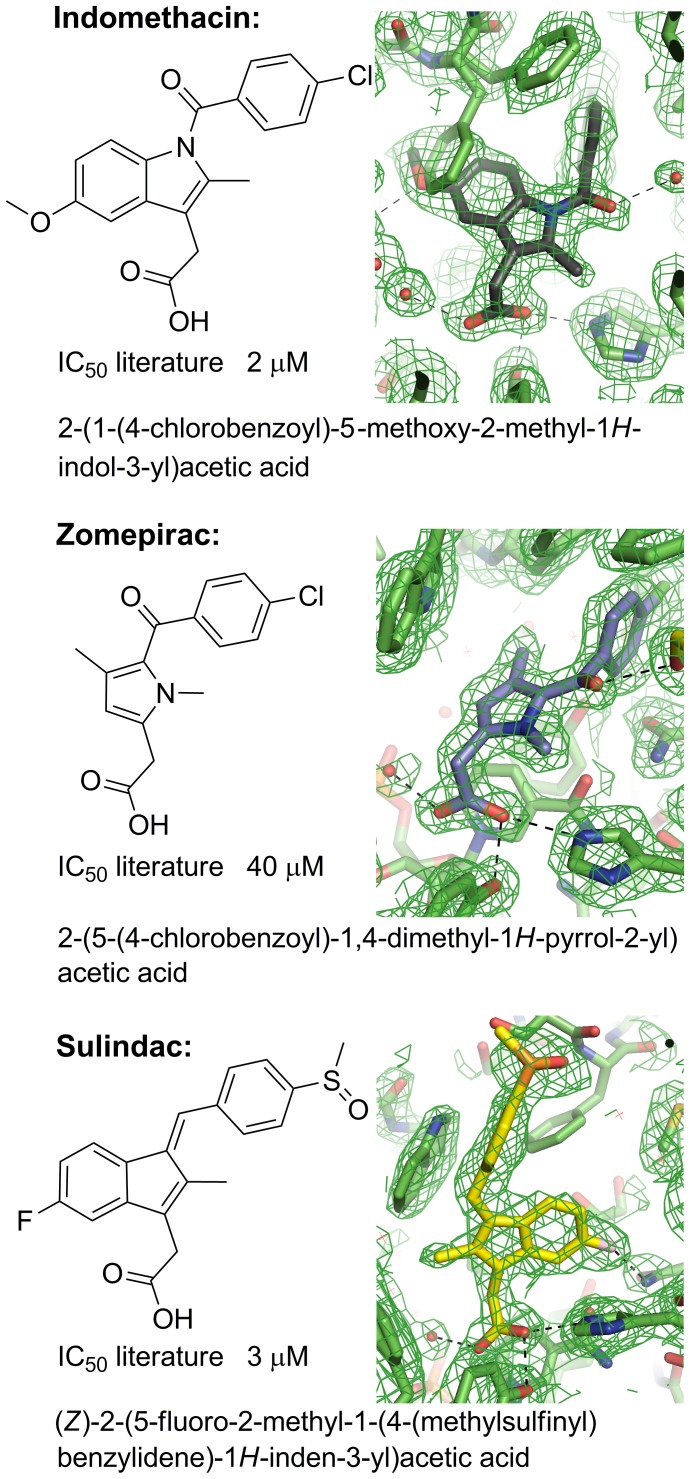
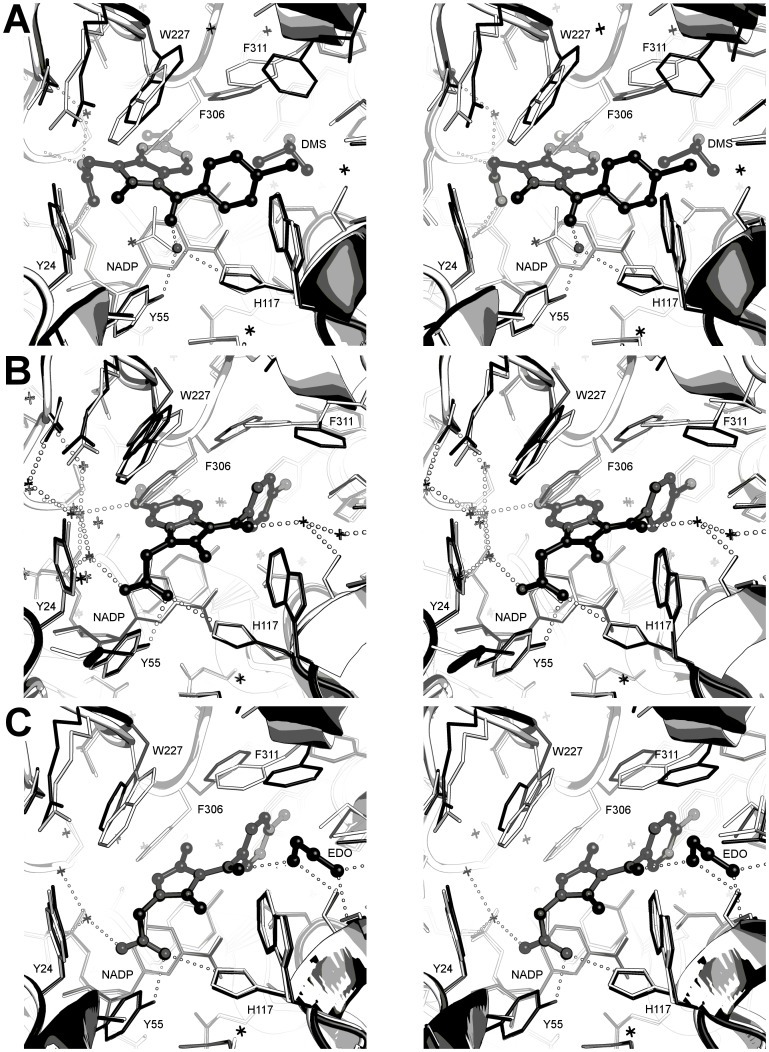
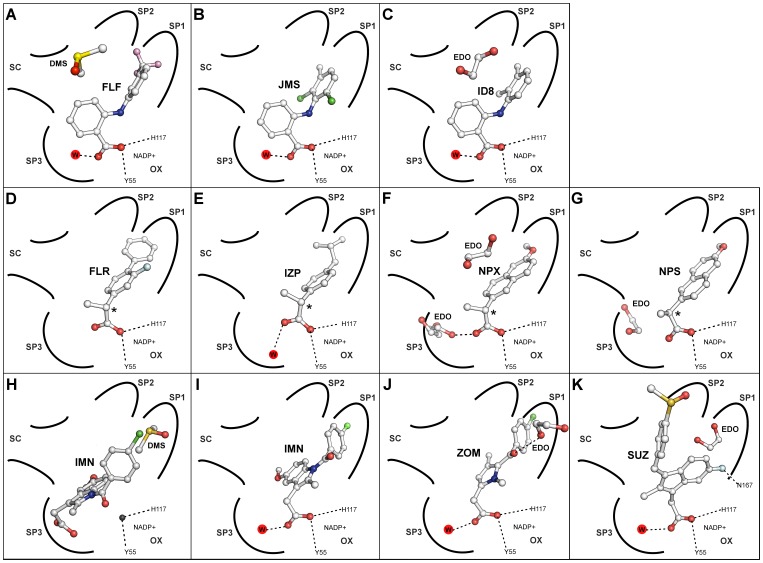
Aldo-keto reductase 1C3 (AKR1C3) catalyses the NADPH dependent reduction of carbonyl groups in a number of important steroid and prostanoid molecules. The enzyme is also over-expressed in prostate and breast cancer and its expression is correlated with the aggressiveness of the disease. The steroid products of AKR1C3 catalysis are important in proliferative signalling of hormone-responsive cells, while the prostanoid products promote prostaglandin-dependent proliferative pathways. In these ways, AKR1C3 contributes to tumour development and maintenance, and suggest that inhibition of AKR1C3 activity is an attractive target for the development of new anti-cancer therapies. Non-steroidal anti-inflammatory drugs (NSAIDs) are one well-known class of compounds that inhibits AKR1C3, yet crystal structures have only been determined for this enzyme with flufenamic acid, indomethacin, and closely related analogues bound. While the flufenamic acid and indomethacin structures have been used to design novel inhibitors, they provide only limited coverage of the NSAIDs that inhibit AKR1C3 and that may be used for the development of new AKR1C3 targeted drugs. To understand how other NSAIDs bind to AKR1C3, we have determined ten crystal structures of AKR1C3 complexes that cover three different classes of NSAID, N-phenylanthranilic acids (meclofenamic acid, mefenamic acid), arylpropionic acids (flurbiprofen, ibuprofen, naproxen), and indomethacin analogues (indomethacin, sulindac, zomepirac). The N-phenylanthranilic and arylpropionic acids bind to common sites including the enzyme catalytic centre and a constitutive active site pocket, with the arylpropionic acids probing the constitutive pocket more effectively. By contrast, indomethacin and the indomethacin analogues sulindac and zomepirac, display three distinctly different binding modes that explain their relative inhibition of the AKR1C family members. This new data from ten crystal structures greatly broadens the base of structures available for future structure-guided drug discovery efforts.
醛酮还原酶 1C3(AKR1C3)催化许多重要的甾体和前列腺素分子中羰基基团的 NADPH 依赖性还原。该酶在前列腺癌和乳腺癌中过度表达,其表达与疾病的侵袭性相关。AKR1C3 催化的甾体产物在激素反应性细胞的增殖信号中很重要,而前列腺素产物则促进前列腺素依赖性增殖途径。通过这些方式,AKR1C3 有助于肿瘤的发展和维持,并表明抑制 AKR1C3 活性是开发新的抗癌疗法的有吸引力的目标。非甾体抗炎药(NSAIDs)是一类众所周知的抑制 AKR1C3 的化合物,然而,只有与 flufenamic acid、indomethacin 和密切相关的类似物结合的情况下,才确定了这种酶的晶体结构。虽然 flufenamic acid 和 indomethacin 的结构已被用于设计新型抑制剂,但它们仅提供了有限的覆盖 AKR1C3 抑制的 NSAIDs 的信息,这些 NSAIDs可能用于开发新的 AKR1C3 靶向药物。为了了解其他 NSAIDs 如何与 AKR1C3 结合,我们已经确定了 AKR1C3 复合物的十个晶体结构,涵盖了三种不同类别的 NSAID,N- 苯基邻氨基苯甲酸(甲芬那酸、甲灭酸)、芳基丙酸(氟比洛芬、布洛芬、萘普生)和 indomethacin 类似物(indomethacin、sulindac、zomepirac)。N- 苯基邻氨基苯甲酸和芳基丙酸结合到共同的位点,包括酶催化中心和组成型活性口袋,芳基丙酸更有效地探测组成型口袋。相比之下,indomethacin 和 indomethacin 类似物 sulindac 和 zomepirac 显示出三种截然不同的结合模式,解释了它们对 AKR1C 家族成员的相对抑制作用。这十个晶体结构的新数据大大扩展了未来基于结构的药物发现工作的结构基础。