Department of Molecular Genetics and Microbiology, Center for Virology, Duke University School of Medicine, Durham, NC 27712, USA.
Cell Host Microbe. 2010 Dec 16;8(6):510-22. doi: 10.1016/j.chom.2010.11.004.
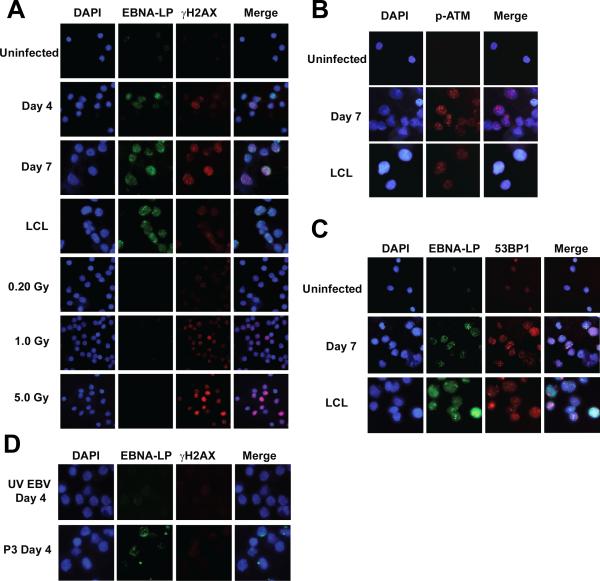
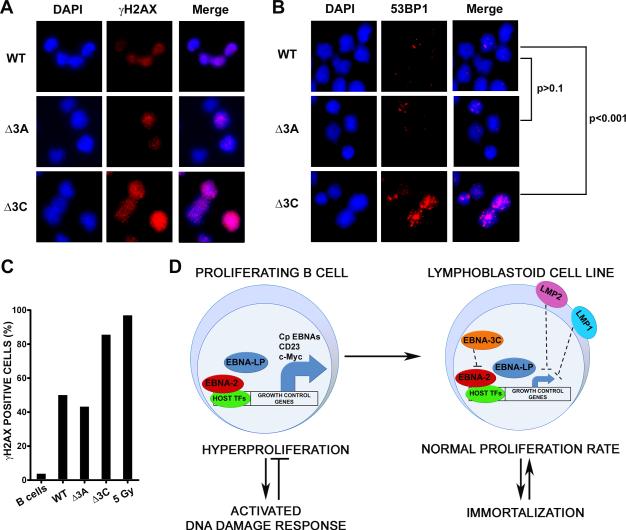
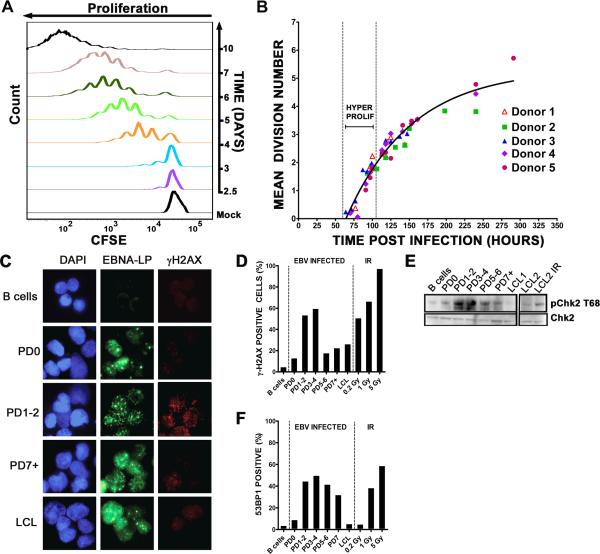
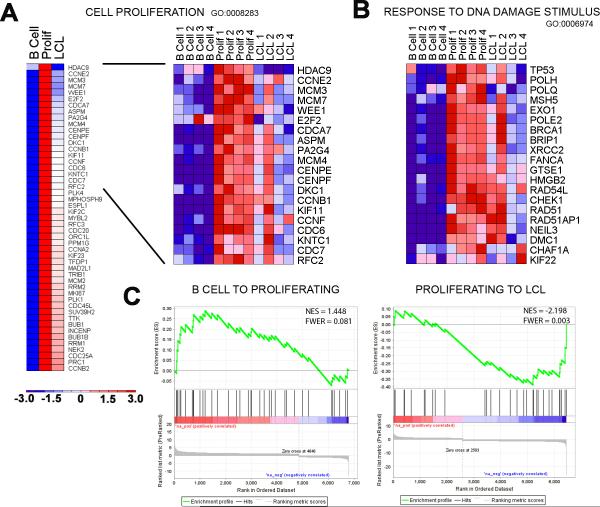
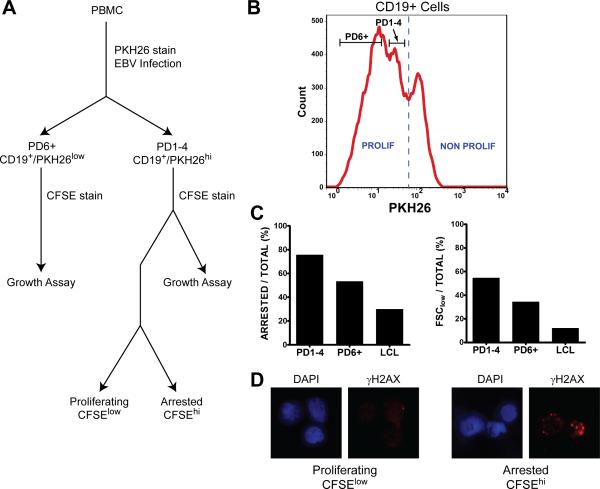
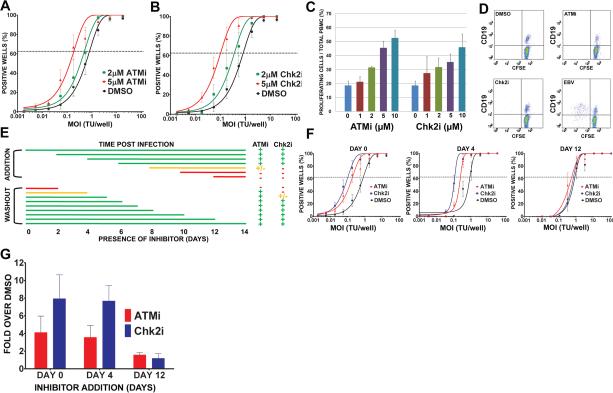
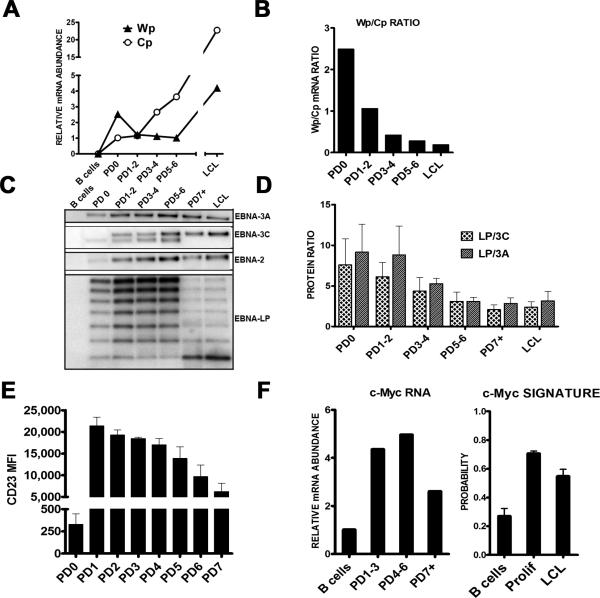
Epstein-Barr virus (EBV), an oncogenic herpesvirus that causes human malignancies, infects and immortalizes primary human B cells in vitro into indefinitely proliferating lymphoblastoid cell lines, which represent a model for EBV-induced tumorigenesis. The immortalization efficiency is very low, suggesting that an innate tumor suppressor mechanism is operative. We identify the DNA damage response (DDR) as a major component of the underlying tumor suppressor mechanism. EBV-induced DDR activation was not due to lytic viral replication, nor did the DDR marks colocalize with latent episomes. Rather, a transient period of EBV-induced hyperproliferation correlated with DDR activation. Inhibition of the DDR kinases ATM and Chk2 markedly increased transformation efficiency of primary B cells. Further, the viral latent oncoprotein EBNA3C was required to attenuate the EBV-induced DDR. We propose that heightened oncogenic activity in early cell divisions activates a growth-suppressive DDR that is attenuated by viral latency products to induce cell immortalization.
EB 病毒(EBV)是一种致癌疱疹病毒,可导致人类恶性肿瘤。它在体外感染并使原代人 B 细胞永生化,形成无限增殖的淋巴母细胞系,这是 EBV 诱导肿瘤发生的模型。永生化效率非常低,这表明存在先天的肿瘤抑制机制。我们发现 DNA 损伤反应(DDR)是潜在肿瘤抑制机制的主要组成部分。EBV 诱导的 DDR 激活不是由于裂解性病毒复制引起的,DDR 标记也没有与潜伏性 EBV episomes 共定位。相反,EBV 诱导的短暂过度增殖期与 DDR 激活相关。DDR 激酶 ATM 和 Chk2 的抑制显著增加了原代 B 细胞的转化效率。此外,病毒潜伏性致癌蛋白 EBNA3C 是减轻 EBV 诱导的 DDR 所必需的。我们提出,早期细胞分裂中增强的致癌活性激活了生长抑制性 DDR,而病毒潜伏产物减轻了 DDR,从而诱导细胞永生化。