Department of Neurology, Massachusetts General Hospital, Charlestown, MA 02129, USA.
Mol Neurodegener. 2010 Dec 14;5:58. doi: 10.1186/1750-1326-5-58.
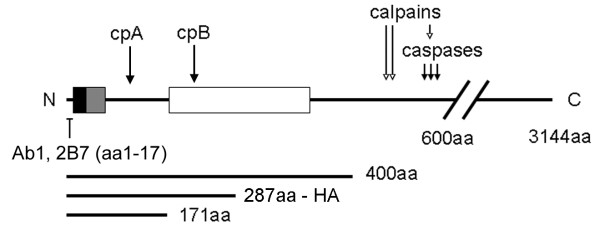
The mutation in Huntington's disease is a polyglutamine expansion near the N-terminus of huntingtin. Huntingtin expressed in immortalized neurons is cleaved near the N-terminus to form N-terminal polypeptides known as cleavage products A and B (cpA and cpB). CpA and cpB with polyglutamine expansion form inclusions in the nucleus and cytoplasm, respectively. The formation of cpA and cpB in primary neurons has not been established and the proteases involved in the formation of these fragments are unknown.
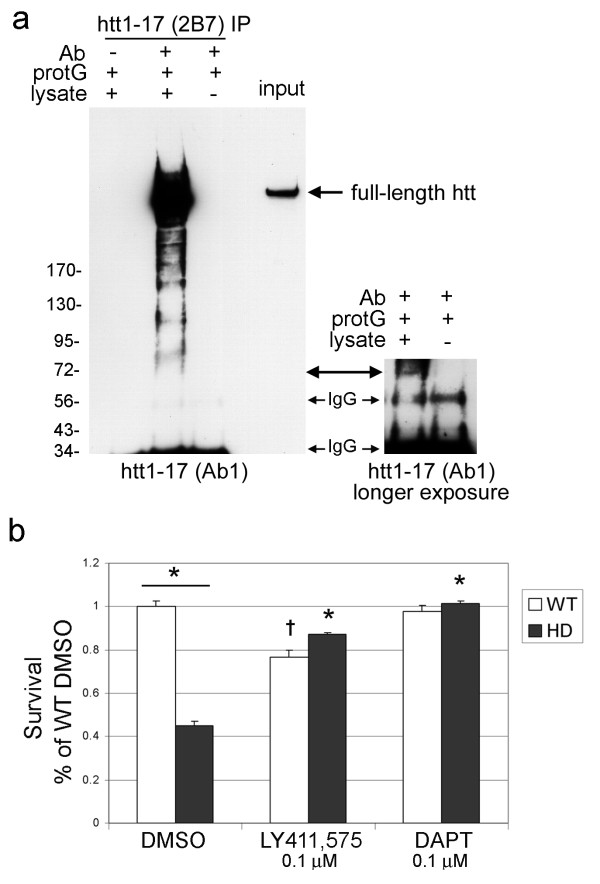
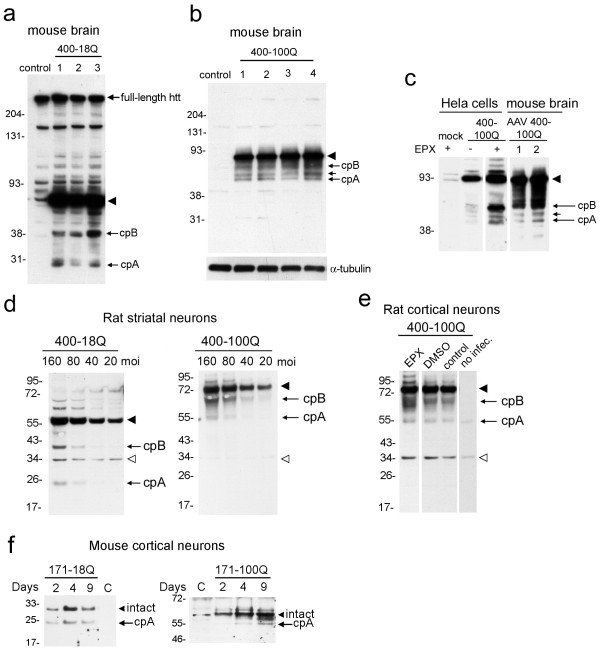
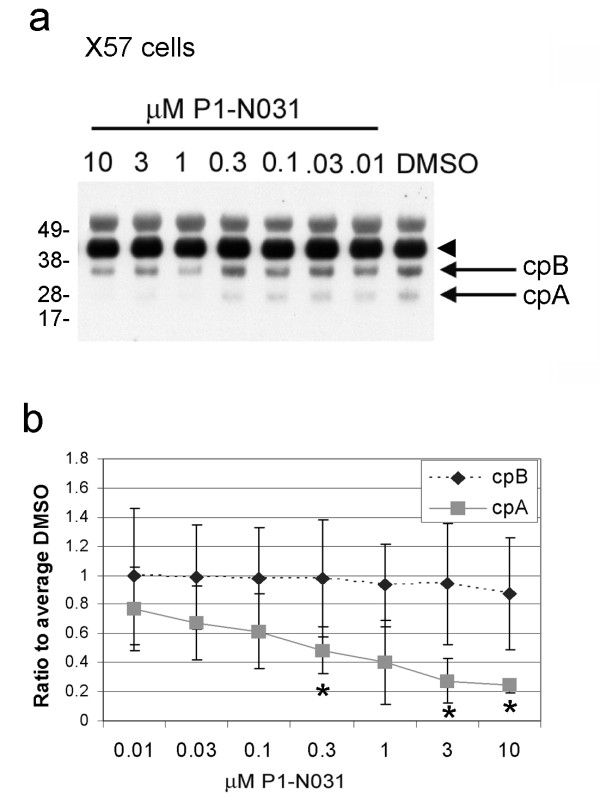
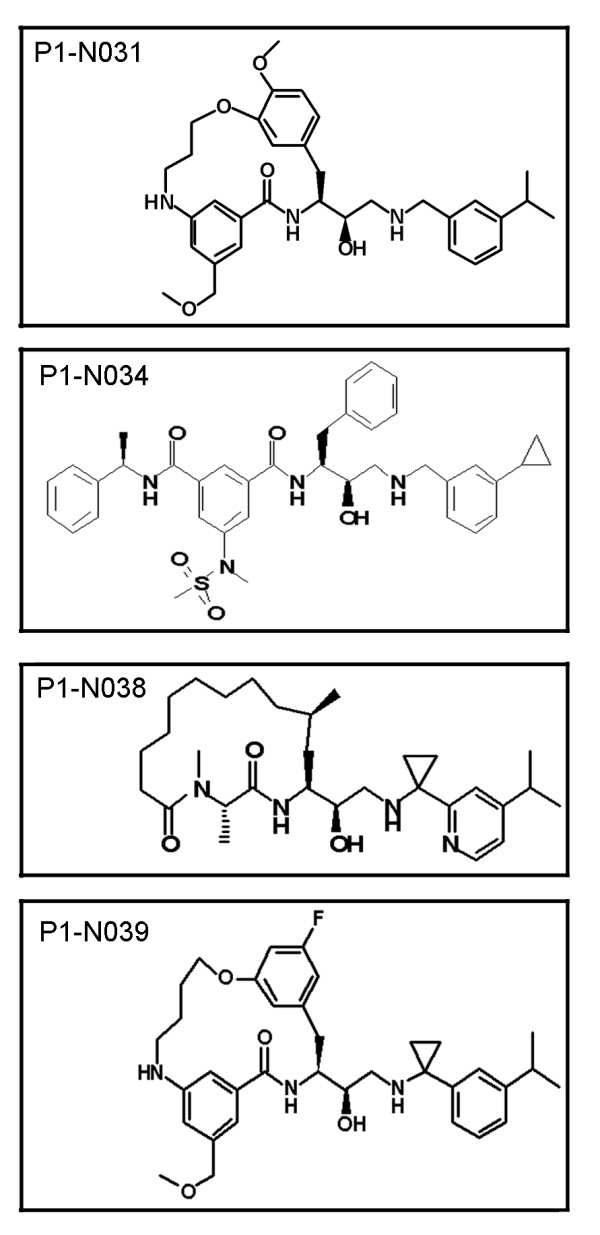
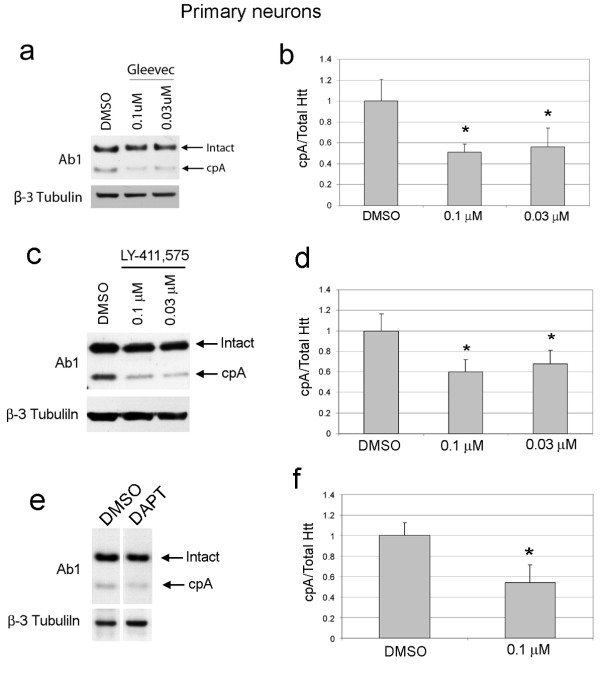
Delivery of htt cDNA into the mouse striatum using adeno-associated virus or into primary cortical neurons using lentivirus generated cpA and cpB, indicating that neurons in brain and in vitro can form these fragments. A screen of small molecule protease inhibitors introduced to clonal striatal X57 cells and HeLa cells identified compounds that reduced levels of cpA and are inhibitors of the aspartyl proteases cathepsin D and cathepsin E. The most effective compound, P1-N031, is a transition state mimetic for aspartyl proteases. By western blot analysis, cathepsin D was easily detected in clonal striatal X57 cells, mouse brain and primary neurons, whereas cathepsin E was only detectible in clonal striatal X57 cells. In primary neurons, levels of cleavage product A were not changed by the same compounds that were effective in clonal striatal cells or by mRNA silencing to partially reduce levels of cathepsin D. Instead, treating primary neurons with compounds that are known to inhibit gamma secretase activity either indirectly (Imatinib mesylate, Gleevec) or selectively (LY-411,575 or DAPT) reduced levels of cpA. LY-411,575 or DAPT also increased survival of primary neurons expressing endogenous full-length mutant huntingtin.
We show that cpA and cpB are produced from a larger huntingtin fragment in vivo in mouse brain and in primary neuron cultures. The aspartyl protease involved in forming cpA has cathepsin-D like properties in immortalized neurons and gamma secretase-like properties in primary neurons, suggesting that cell type may be a critical factor that specifies the aspartyl protease responsible for cpA. Since gamma secretase inhibitors were also protective in primary neurons, further study of the role of gamma-secretase activity in HD neurons is justified.
亨廷顿病的突变是位于亨廷顿蛋白(huntingtin)N 端附近的多聚谷氨酰胺扩展。在永生化神经元中表达的亨廷顿蛋白在 N 端附近被切割,形成称为切割产物 A 和 B(cpA 和 cpB)的 N 端多肽。具有多聚谷氨酰胺扩展的 cpA 和 cpB 分别在核和细胞质中形成包含物。在原代神经元中尚未建立 cpA 和 cpB 的形成,并且参与这些片段形成的蛋白酶尚不清楚。
使用腺相关病毒将 htt cDNA 递送至小鼠纹状体或使用慢病毒将其递送至原代皮质神经元,生成 cpA 和 cpB,表明脑内和体外神经元可以形成这些片段。对克隆纹状体 X57 细胞和 HeLa 细胞引入的小分子蛋白酶抑制剂进行筛选,鉴定出降低 cpA 水平的化合物,并且是天冬氨酸蛋白酶组织蛋白酶 D 和组织蛋白酶 E 的抑制剂。最有效的化合物 P1-N031 是天冬氨酸蛋白酶的过渡态模拟物。通过 Western blot 分析,在克隆纹状体 X57 细胞、小鼠脑和原代神经元中容易检测到组织蛋白酶 D,而仅在克隆纹状体 X57 细胞中检测到组织蛋白酶 E。在原代神经元中,相同的化合物对 cpA 水平没有影响,这些化合物在克隆纹状体细胞中有效,或通过 mRNA 沉默部分降低组织蛋白酶 D 水平。相反,用已知间接(伊马替尼甲磺酸盐,格列卫)或选择性(LY-411,575 或 DAPT)抑制γ分泌酶活性的化合物处理原代神经元可降低 cpA 水平。LY-411,575 或 DAPT 还增加了表达内源性全长突变型亨廷顿蛋白的原代神经元的存活率。
我们表明,cpA 和 cpB 是在体内从更大的亨廷顿蛋白片段在小鼠脑和原代神经元培养物中产生的。参与形成 cpA 的天冬氨酸蛋白酶在永生化神经元中具有组织蛋白酶 D 样特性,在原代神经元中具有 γ 分泌酶样特性,这表明细胞类型可能是决定负责 cpA 的天冬氨酸蛋白酶的关键因素。由于 γ 分泌酶抑制剂在原代神经元中也具有保护作用,因此进一步研究 γ-分泌酶活性在 HD 神经元中的作用是合理的。