Gu Fei, Hsu Hang-Kai, Hsu Pei-Yin, Wu Jiejun, Ma Yilin, Parvin Jeffrey, Huang Tim H-M, Jin Victor X
Department of Biomedical Informatics, The Ohio State University, Columbus, USA.
BMC Syst Biol. 2010 Dec 17;4:170. doi: 10.1186/1752-0509-4-170.
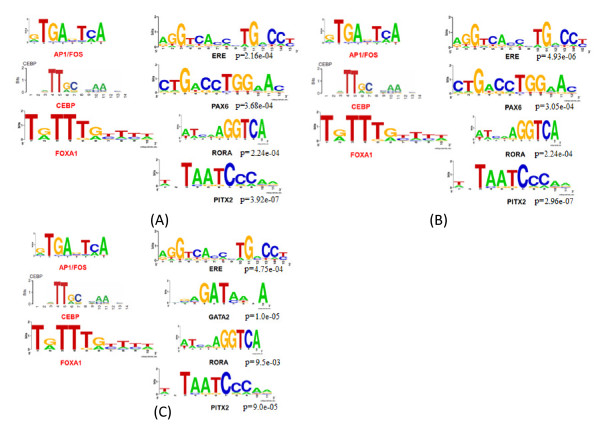
Global profiling of in vivo protein-DNA interactions using ChIP-based technologies has evolved rapidly in recent years. Although many genome-wide studies have identified thousands of ERα binding sites and have revealed the associated transcription factor (TF) partners, such as AP1, FOXA1 and CEBP, little is known about ERα associated hierarchical transcriptional regulatory networks.
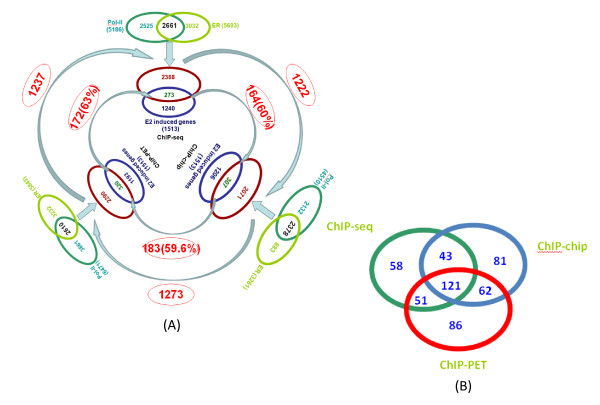
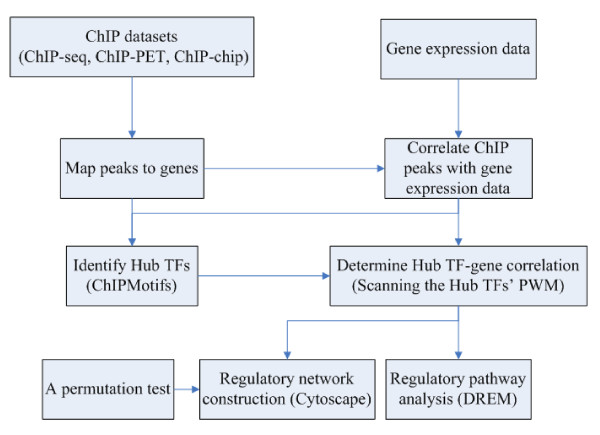
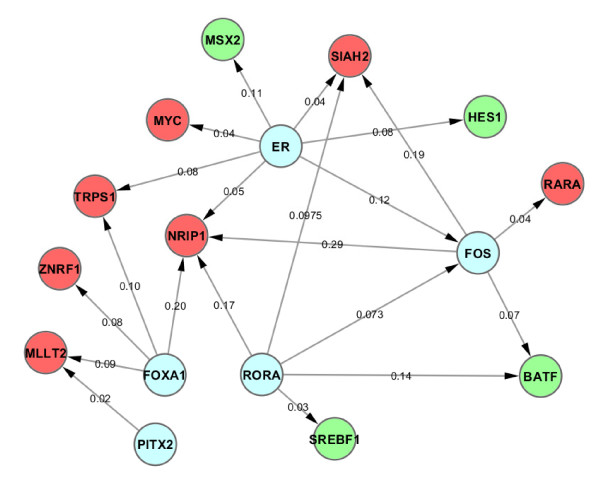
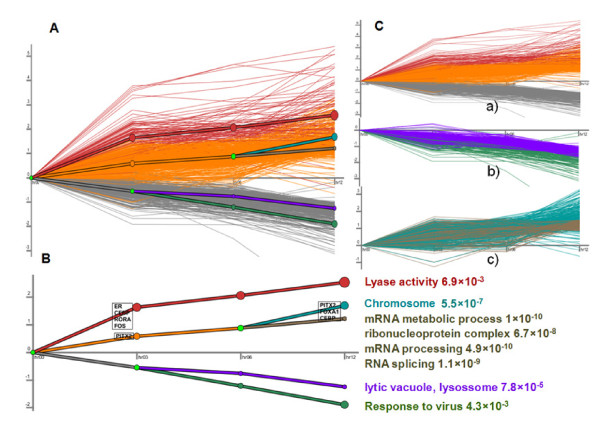
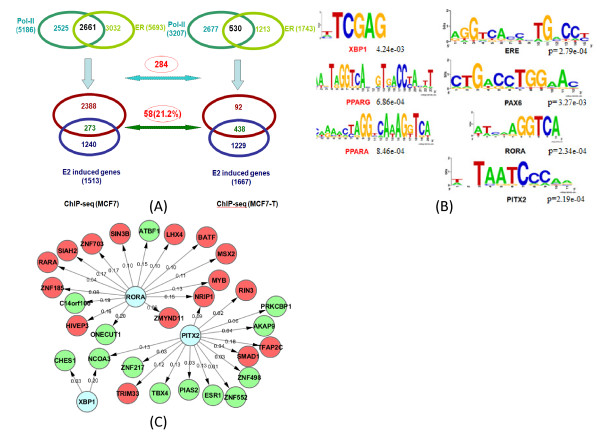
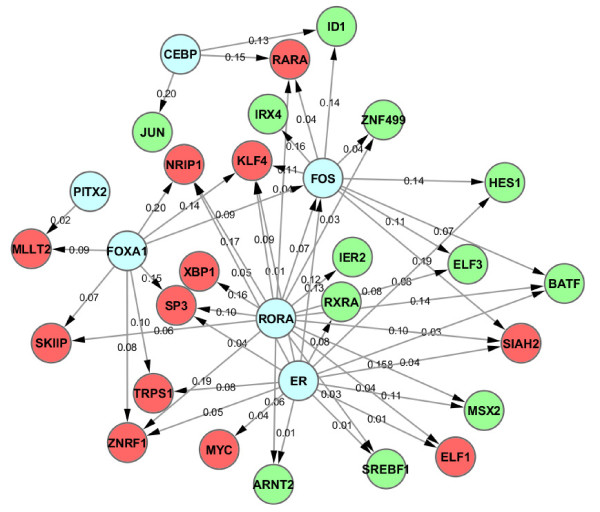
In this study, we applied computational approaches to analyze three public available ChIP-based datasets: ChIP-seq, ChIP-PET and ChIP-chip, and to investigate the hierarchical regulatory network for ERα and ERα partner TFs regulation in estrogen-dependent breast cancer MCF7 cells. 16 common TFs and two common new TF partners (RORA and PITX2) were found among ChIP-seq, ChIP-chip and ChIP-PET datasets. The regulatory networks were constructed by scanning the ChIP-peak region with TF specific position weight matrix (PWM). A permutation test was performed to test the reliability of each connection of the network. We then used DREM software to perform gene ontology function analysis on the common genes. We found that FOS, PITX2, RORA and FOXA1 were involved in the up-regulated genes.We also conducted the ERα and Pol-II ChIP-seq experiments in tamoxifen resistance MCF7 cells (denoted as MCF7-T in this study) and compared the difference between MCF7 and MCF7-T cells. The result showed very little overlap between these two cells in terms of targeted genes (21.2% of common genes) and targeted TFs (25% of common TFs). The significant dissimilarity may indicate totally different transcriptional regulatory mechanisms between these two cancer cells.
Our study uncovers new estrogen-mediated regulatory networks by mining three ChIP-based data in MCF7 cells and ChIP-seq data in MCF7-T cells. We compared the different ChIP-based technologies as well as different breast cancer cells. Our computational analytical approach may guide biologists to further study the underlying mechanisms in breast cancer cells or other human diseases.
近年来,基于染色质免疫沉淀(ChIP)技术的体内蛋白质 - DNA相互作用的全局分析发展迅速。尽管许多全基因组研究已经鉴定出数千个雌激素受体α(ERα)结合位点,并揭示了相关的转录因子(TF)伙伴,如活化蛋白1(AP1)、叉头框蛋白A1(FOXA1)和CCAAT增强子结合蛋白(CEBP),但对于ERα相关的层次转录调控网络却知之甚少。
在本研究中,我们应用计算方法分析了三个公开可用的基于ChIP的数据集:ChIP测序(ChIP-seq)、ChIP成对末端标签测序(ChIP-PET)和ChIP芯片(ChIP-chip),以研究雌激素依赖性乳腺癌MCF7细胞中ERα及其伙伴TFs调控的层次调控网络。在ChIP-seq、ChIP-chip和ChIP-PET数据集中发现了16个常见的TFs和两个常见的新TF伙伴(视黄酸相关孤儿受体A,RORA;垂体特异性转录因子2,PITX2)。通过用TF特异性位置权重矩阵(PWM)扫描ChIP峰区域构建调控网络。进行置换检验以测试网络中每个连接的可靠性。然后我们使用DREM软件对共同基因进行基因本体功能分析。我们发现FOS、PITX2、RORA和FOXA1参与了上调基因。我们还在他莫昔芬耐药的MCF7细胞(本研究中记为MCF7-T)中进行了ERα和RNA聚合酶II(Pol-II)的ChIP-seq实验,并比较了MCF7和MCF7-T细胞之间的差异。结果表明,这两种细胞在靶向基因(共同基因的21.2%)和靶向TFs(共同TFs的25%)方面重叠很少。这种显著差异可能表明这两种癌细胞之间存在完全不同的转录调控机制。
我们的研究通过挖掘MCF7细胞中的三个基于ChIP的数据和MCF7-T细胞中的ChIP-seq数据,揭示了新的雌激素介导的调控网络。我们比较了不同的基于ChIP的技术以及不同的乳腺癌细胞。我们的计算分析方法可能会指导生物学家进一步研究乳腺癌细胞或其他人类疾病的潜在机制。