University of Maryland Greenebaum Cancer Center, 22 South Greene Street, Baltimore, MD 21201, USA.
BMC Bioinformatics. 2010 Dec 21;11:606. doi: 10.1186/1471-2105-11-606.
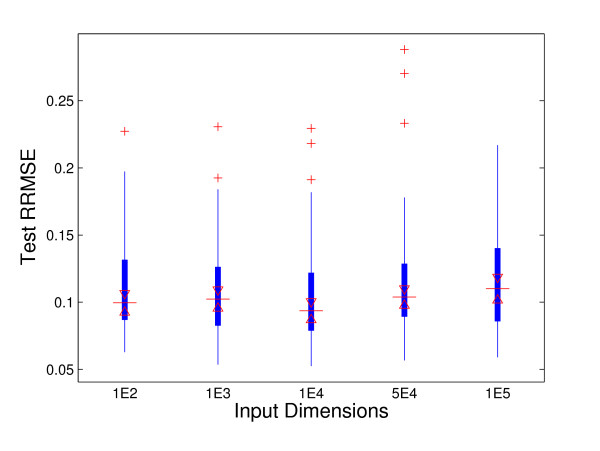
Most genomic data have ultra-high dimensions with more than 10,000 genes (probes). Regularization methods with L₁ and L(p) penalty have been extensively studied in survival analysis with high-dimensional genomic data. However, when the sample size n << m (the number of genes), directly identifying a small subset of genes from ultra-high (m > 10, 000) dimensional data is time-consuming and not computationally efficient. In current microarray analysis, what people really do is select a couple of thousands (or hundreds) of genes using univariate analysis or statistical tests, and then apply the LASSO-type penalty to further reduce the number of disease associated genes. This two-step procedure may introduce bias and inaccuracy and lead us to miss biologically important genes.
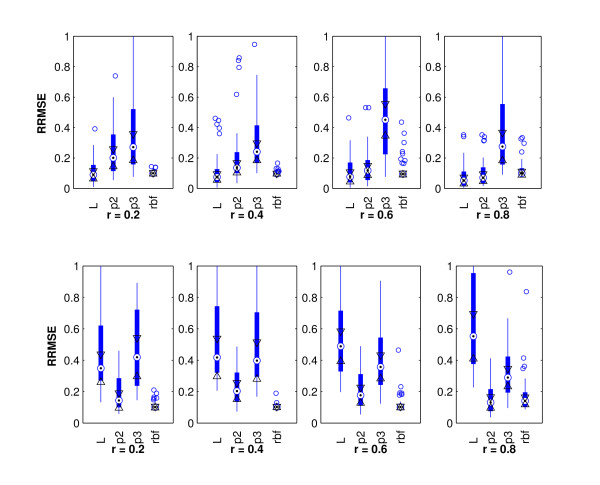
The accelerated failure time (AFT) model is a linear regression model and a useful alternative to the Cox model for survival analysis. In this paper, we propose a nonlinear kernel based AFT model and an efficient variable selection method with adaptive kernel ridge regression. Our proposed variable selection method is based on the kernel matrix and dual problem with a much smaller n x n matrix. It is very efficient when the number of unknown variables (genes) is much larger than the number of samples. Moreover, the primal variables are explicitly updated and the sparsity in the solution is exploited.
Our proposed methods can simultaneously identify survival associated prognostic factors and predict survival outcomes with ultra-high dimensional genomic data. We have demonstrated the performance of our methods with both simulation and real data. The proposed method performs superbly with limited computational studies.
大多数基因组数据具有超过 10000 个基因(探针)的超高维特性。在高维基因组数据的生存分析中,已经广泛研究了具有 L₁和 L(p)惩罚的正则化方法。然而,当样本量 n << m(基因数量)时,直接从超高维(m>10000)数据中识别一小部分基因是非常耗时的,并且计算效率不高。在当前的微阵列分析中,人们真正做的是使用单变量分析或统计检验选择几千个(或几百个)基因,然后应用 LASSO 型惩罚进一步减少与疾病相关的基因数量。这种两步程序可能会引入偏差和不准确性,导致我们错过生物学上重要的基因。
加速失效时间(AFT)模型是一种线性回归模型,是生存分析中 Cox 模型的有用替代方法。在本文中,我们提出了一种基于非线性核的 AFT 模型和一种基于自适应核岭回归的高效变量选择方法。我们提出的变量选择方法基于核矩阵和对偶问题,使用的 n x n 矩阵要小得多。当未知变量(基因)的数量远大于样本数量时,它的效率非常高。此外,还显式更新了主变量,并利用了解中的稀疏性。
我们提出的方法可以同时识别与生存相关的预后因素,并利用超高维基因组数据预测生存结果。我们已经通过模拟和真实数据验证了我们方法的性能。该方法在有限的计算研究中表现出色。