Shahani Vijay M, Yue Peibin, Haftchenary Sina, Zhao Wei, Lukkarila Julie L, Zhang Xiaolei, Ball Daniel, Nona Christina, Gunning Patrick T, Turkson James
ACS Med Chem Lett. 2011 Jan 13;2(1):79-84. doi: 10.1021/ml100224d. Epub 2010 Oct 25.
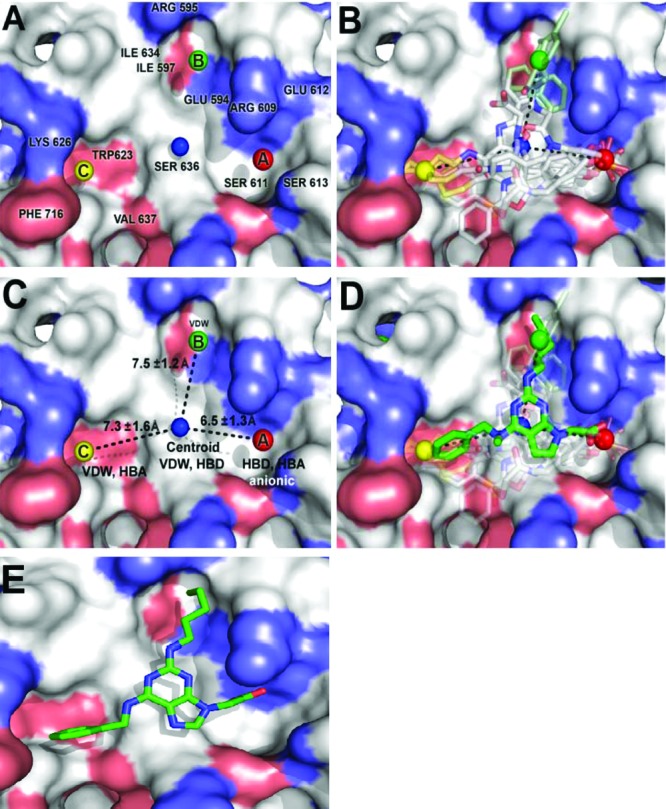
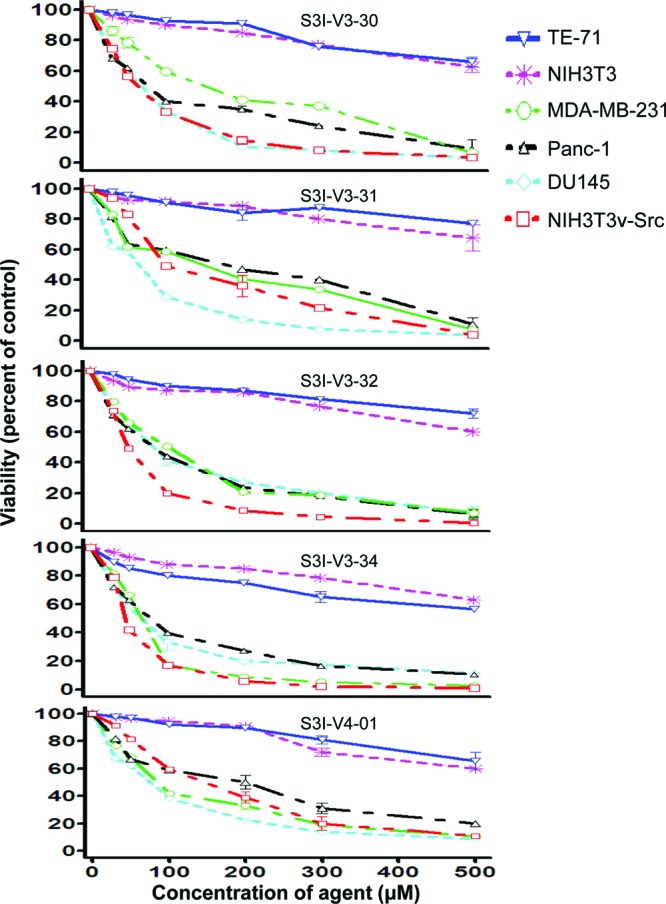
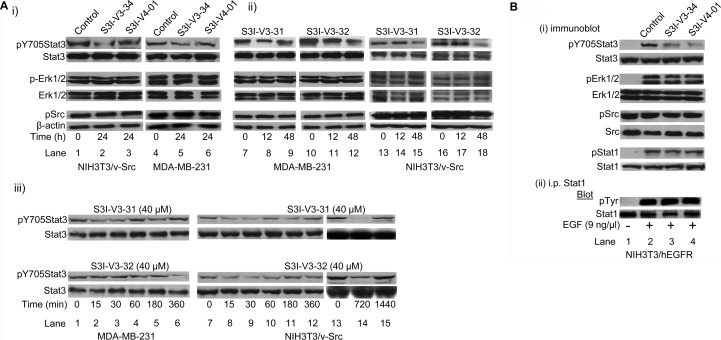
To facilitate the discovery of clinically useful Stat3 inhibitors, computational analysis of the binding to Stat3 of the existing Stat3 dimerization disruptors and quantitative structure-activity relationships (QSAR) were pursued, by which a pharmacophore model was derived for predicting optimized Stat3 dimerization inhibitors. The 2,6,9-trisubstituted-purine scaffold was functionalized in order to access the three subpockets of the Stat3 SH2 domain surface and to derive potent Stat3-binding inhibitors. Select purine scaffolds showed good affinities (K(D), 0.8-12 μM) for purified, nonphosphorylated Stat3 and inhibited Stat3 DNA-binding activity in vitro and intracellular phosphorylation at 20-60 μM. Furthermore, agents selectively suppressed viability of human prostate, breast and pancreatic cancer cells, and v-Src-transformed mouse fibroblasts that harbor aberrant Stat3 activity. Studies herein identified novel small-molecule trisubstituted purines as effective inhibitors of constitutively active Stat3 and of the viability of Stat3-dependent tumor cells, and are the first to validate the use of purine bases as templates for building novel Stat3 inhibitors.
为了促进临床可用的Stat3抑制剂的发现,我们对现有Stat3二聚化破坏剂与Stat3的结合进行了计算分析,并研究了定量构效关系(QSAR),由此推导出一个药效团模型,用于预测优化的Stat3二聚化抑制剂。对2,6,9-三取代嘌呤骨架进行功能化修饰,以便进入Stat3 SH2结构域表面的三个亚口袋,并获得强效的Stat3结合抑制剂。部分嘌呤骨架对纯化的非磷酸化Stat3显示出良好的亲和力(K(D),0.8 - 12 μM),并在20 - 60 μM浓度下抑制体外Stat3 DNA结合活性和细胞内磷酸化。此外,这些试剂选择性地抑制了人前列腺癌、乳腺癌和胰腺癌细胞以及具有异常Stat3活性的v-Src转化小鼠成纤维细胞的活力。本研究确定了新型小分子三取代嘌呤作为组成型激活Stat3和Stat3依赖性肿瘤细胞活力的有效抑制剂,并且首次验证了使用嘌呤碱基作为构建新型Stat3抑制剂的模板。