Gregor Mendel Institute, Vienna, Austria.
PLoS One. 2011 Jan 5;6(1):e15292. doi: 10.1371/journal.pone.0015292.
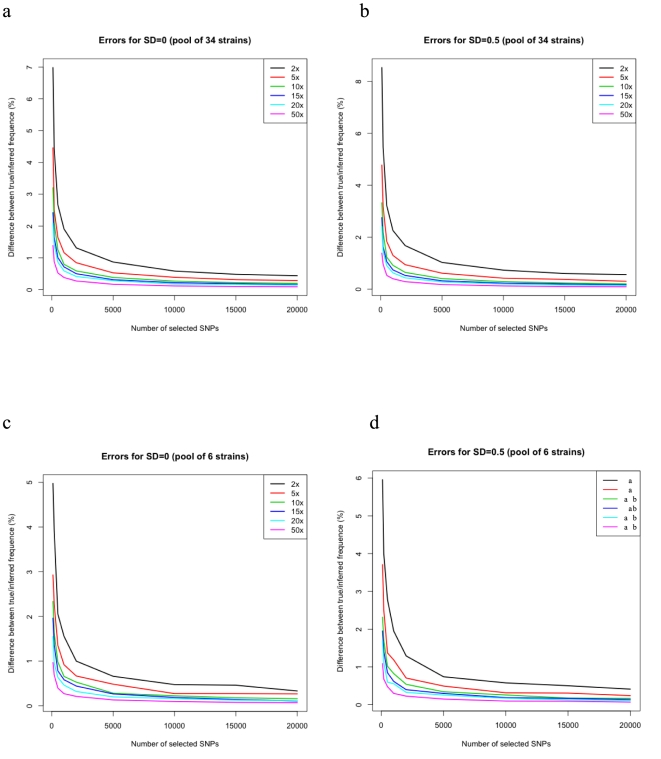
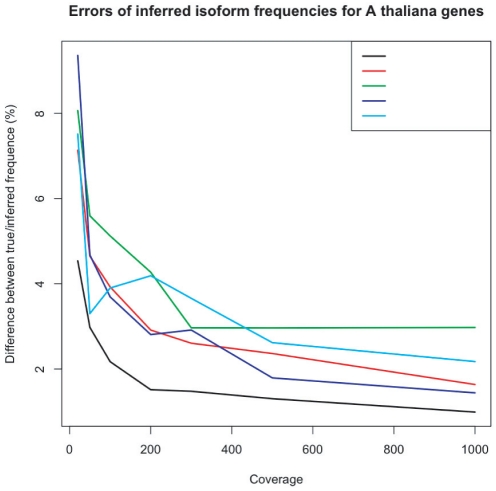
With the advance of next-generation sequencing (NGS) technologies, increasingly ambitious applications are becoming feasible. A particularly powerful one is the sequencing of polymorphic, pooled samples. The pool can be naturally occurring, as in the case of multiple pathogen strains in a blood sample, multiple types of cells in a cancerous tissue sample, or multiple isoforms of mRNA in a cell. In these cases, it's difficult or impossible to partition the subtypes experimentally before sequencing, and those subtype frequencies must hence be inferred. In addition, investigators may occasionally want to artificially pool the sample of a large number of individuals for reasons of cost-efficiency, e.g., when carrying out genetic mapping using bulked segregant analysis. Here we describe PoolHap, a computational tool for inferring haplotype frequencies from pooled samples when haplotypes are known. The key insight into why PoolHap works is that the large number of SNPs that come with genome-wide coverage can compensate for the uneven coverage across the genome. The performance of PoolHap is illustrated and discussed using simulated and real data. We show that PoolHap is able to accurately estimate the proportions of haplotypes with less than 2% error for 34-strain mixtures with 2X total coverage Arabidopsis thaliana whole genome polymorphism data. This method should facilitate greater biological insight into heterogeneous samples that are difficult or impossible to isolate experimentally. Software and users manual are freely available at http://arabidopsis.gmi.oeaw.ac.at/quan/poolhap/.
随着下一代测序(NGS)技术的进步,越来越多雄心勃勃的应用变得可行。其中一个特别强大的应用是对多态性、混合样本进行测序。该池可以是自然发生的,例如在血液样本中存在多种病原体菌株、在癌组织样本中存在多种类型的细胞、或在细胞中存在多种 mRNA 同工型的情况下。在这些情况下,在测序之前很难或不可能通过实验对亚类进行分区,因此必须推断这些亚类频率。此外,研究人员可能偶尔出于成本效益的原因希望人工混合大量个体的样本,例如,当使用 bulked segregant analysis 进行遗传图谱绘制时。在这里,我们描述了 PoolHap,这是一种当已知单倍型时从混合样本中推断单倍型频率的计算工具。PoolHap 之所以有效的关键见解是,基因组覆盖范围内大量的 SNP 可以弥补基因组覆盖不均的问题。使用模拟和真实数据说明了和讨论了 PoolHap 的性能。我们表明,PoolHap 能够以小于 2%的误差准确估计具有 2X 总覆盖度的拟南芥全基因组多态性数据的 34 株混合物的单倍型比例。该方法应该有助于更好地了解难以或不可能通过实验分离的异质样本的生物学特性。软件和用户手册可在 http://arabidopsis.gmi.oeaw.ac.at/quan/poolhap/ 免费获得。