Cuccato Giulia, Polynikis Athanasios, Siciliano Velia, Graziano Mafalda, di Bernardo Mario, di Bernardo Diego
Telethon Institute of Genetics and Medicine (TIGEM), Naples, Italy.
BMC Syst Biol. 2011 Jan 27;5:19. doi: 10.1186/1752-0509-5-19.
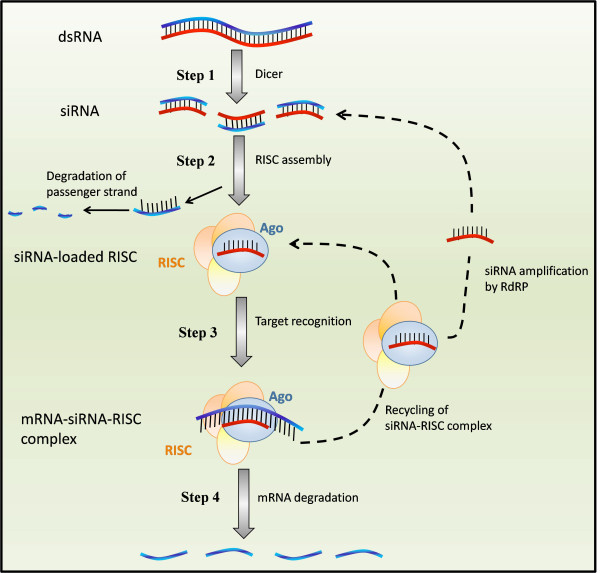
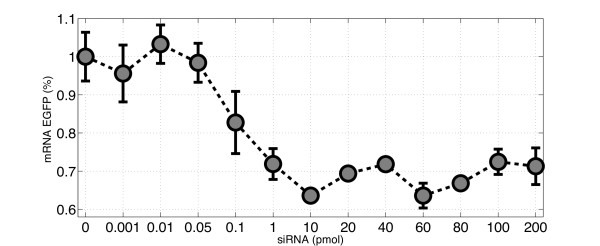
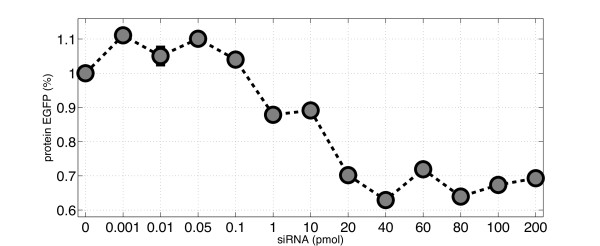
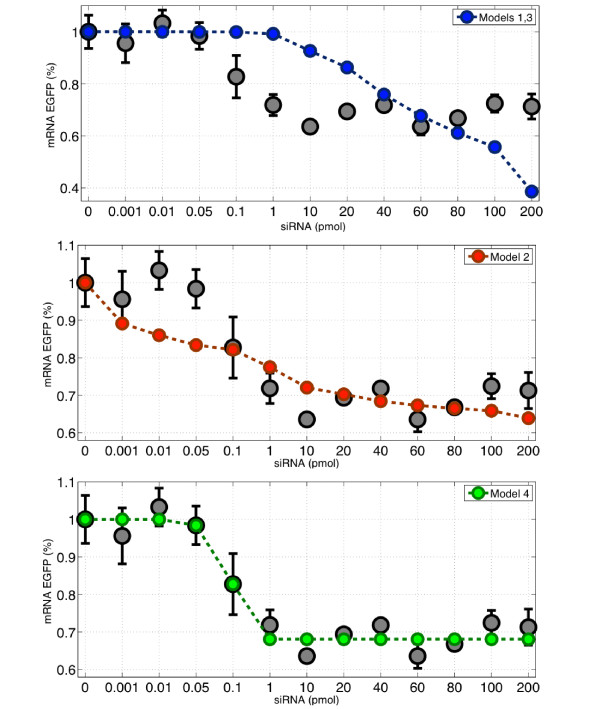
RNA interference (RNAi) is a regulatory cellular process that controls post-transcriptional gene silencing. During RNAi double-stranded RNA (dsRNA) induces sequence-specific degradation of homologous mRNA via the generation of smaller dsRNA oligomers of length between 21-23nt (siRNAs). siRNAs are then loaded onto the RNA-Induced Silencing multiprotein Complex (RISC), which uses the siRNA antisense strand to specifically recognize mRNA species which exhibit a complementary sequence. Once the siRNA loaded-RISC binds the target mRNA, the mRNA is cleaved and degraded, and the siRNA loaded-RISC can degrade additional mRNA molecules. Despite the widespread use of siRNAs for gene silencing, and the importance of dosage for its efficiency and to avoid off target effects, none of the numerous mathematical models proposed in literature was validated to quantitatively capture the effects of RNAi on the target mRNA degradation for different concentrations of siRNAs. Here, we address this pressing open problem performing in vitro experiments of RNAi in mammalian cells and testing and comparing different mathematical models fitting experimental data to in-silico generated data. We performed in vitro experiments in human and hamster cell lines constitutively expressing respectively EGFP protein or tTA protein, measuring both mRNA levels, by quantitative Real-Time PCR, and protein levels, by FACS analysis, for a large range of concentrations of siRNA oligomers.
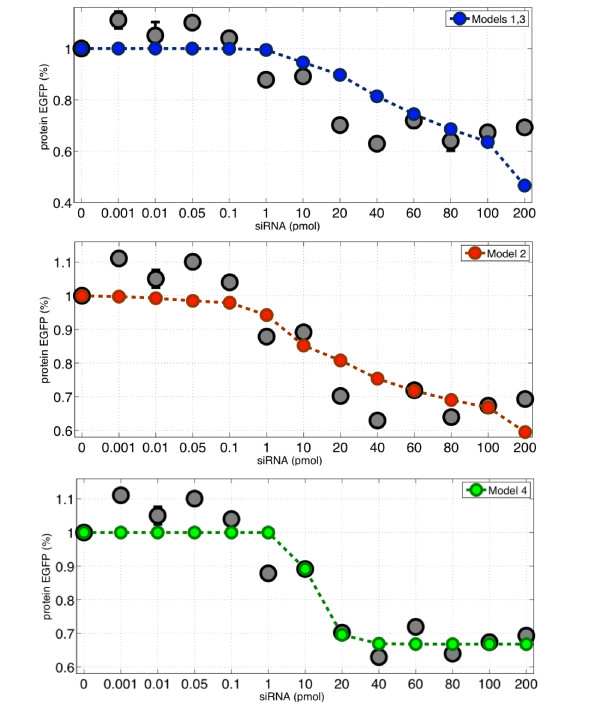
We tested and validated four different mathematical models of RNA interference by quantitatively fitting models' parameters to best capture the in vitro experimental data. We show that a simple Hill kinetic model is the most efficient way to model RNA interference. Our experimental and modeling findings clearly show that the RNAi-mediated degradation of mRNA is subject to saturation effects.
Our model has a simple mathematical form, amenable to analytical investigations and a small set of parameters with an intuitive physical meaning, that makes it a unique and reliable mathematical tool. The findings here presented will be a useful instrument for better understanding RNAi biology and as modelling tool in Systems and Synthetic Biology.
RNA干扰(RNAi)是一种调控细胞过程,可控制转录后基因沉默。在RNAi过程中,双链RNA(dsRNA)通过生成长度在21 - 23个核苷酸之间的较小dsRNA寡聚物(小干扰RNA,siRNAs)诱导同源mRNA的序列特异性降解。然后,siRNAs被加载到RNA诱导沉默多蛋白复合体(RISC)上,该复合体利用siRNA反义链特异性识别具有互补序列的mRNA种类。一旦加载了siRNA的RISC与靶mRNA结合,mRNA就会被切割和降解,并且加载了siRNA的RISC可以降解其他mRNA分子。尽管siRNAs在基因沉默中被广泛使用,且剂量对其效率和避免脱靶效应很重要,但文献中提出的众多数学模型均未经过验证,无法定量捕捉不同浓度siRNAs对RNAi介导的靶mRNA降解的影响。在此,我们通过在哺乳动物细胞中进行RNAi的体外实验,并测试和比较将不同数学模型与实验数据拟合到计算机生成数据的情况,来解决这个紧迫的开放性问题。我们在分别组成型表达绿色荧光蛋白(EGFP)或四环素反式激活蛋白(tTA)的人源和仓鼠细胞系中进行了体外实验,通过定量实时PCR测量mRNA水平,并通过流式细胞术分析测量了大范围浓度的siRNA寡聚物的蛋白质水平。
我们通过将模型参数定量拟合以最佳捕捉体外实验数据,测试并验证了四种不同的RNA干扰数学模型。我们表明,简单的希尔动力学模型是模拟RNA干扰的最有效方法。我们的实验和建模结果清楚地表明,RNAi介导的mRNA降解存在饱和效应。
我们的模型具有简单的数学形式,适合进行分析研究,且参数集小且具有直观的物理意义,这使其成为一种独特且可靠的数学工具。此处呈现的研究结果将成为更好理解RNAi生物学以及作为系统与合成生物学建模工具的有用手段。