Service de Neurologie, CHU de Saint-Etienne, Saint-Etienne, France.
Orphanet J Rare Dis. 2011 Feb 5;6:4. doi: 10.1186/1750-1172-6-4.
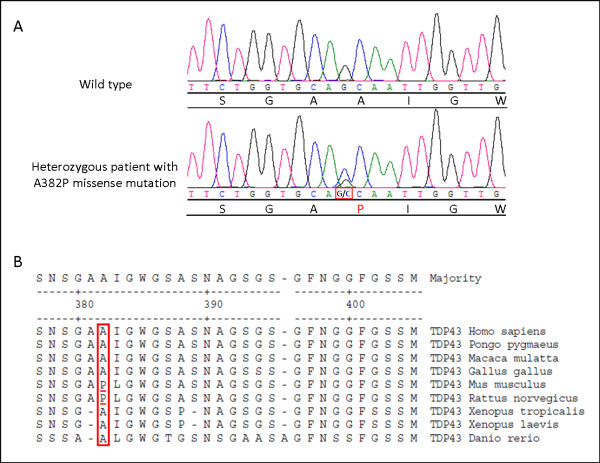
Patients with TARDBP mutations have so far been classified as ALS, sometimes with frontal lobe dysfunction. A 66-year-old patient progressively developed a severe sensory disorder, followed by a motor disorder, which evolved over nine years. Symptoms started in the left hand and slowly involved the four limbs. Investigations were consistent with a mixed sensory and motor neuronopathy. A heterozygous change from an alanine to a proline at amino acid 382 was identified in exon 6 of the TARDPB gene (p.A382P). This case expands the phenotypic spectrum associated with mutations in the TARDBP gene and shows that sensory neurons can be severely damaged early in the course of the disease, following a propagating process, with an orderly progression from a focal starting point. A combination of severe sensory and motor neuronopathy is rarely encountered in clinical practice. The possibility of an A382P TDP-43 mutation should be considered in patients with such an association.
患者携带 TARDBP 突变,目前被归类为 ALS,有时伴有额叶功能障碍。一位 66 岁的患者逐渐出现严重的感觉障碍,随后出现运动障碍,病程长达九年。症状从左手开始,缓慢累及四肢。检查结果符合混合性感觉运动神经元病。在 TARDPB 基因的外显子 6 中发现了一个从丙氨酸到脯氨酸的杂合性变化,即 382 位氨基酸(p.A382P)。该病例扩展了与 TARDBP 基因突变相关的表型谱,并表明感觉神经元可能在疾病早期沿着传播过程受到严重损害,从一个局灶性起始点有序进展。在临床实践中很少遇到严重的感觉运动神经元病同时存在的情况。在出现这种关联的患者中,应考虑是否存在 A382P TDP-43 突变。