Department of Biochemistry and Molecular Genetics, University of Colorado School of Medicine, Aurora, CO, USA.
Hum Genomics. 2011 Jan;5(2):117-23. doi: 10.1186/1479-7364-5-2-117.
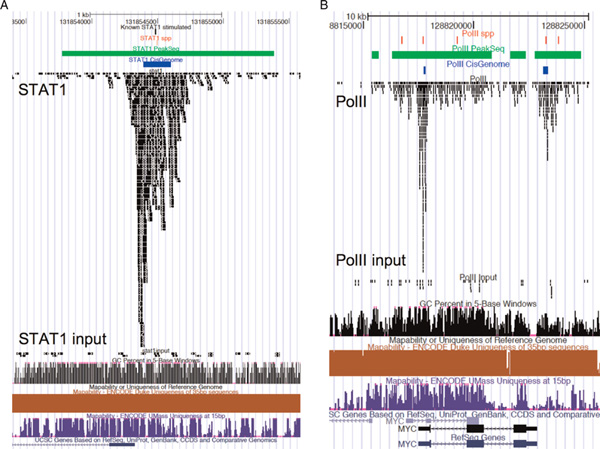
Chromatin immunoprecipitation followed by massively parallel next-generation sequencing (ChIP-seq) is a valuable experimental strategy for assaying protein-DNA interaction over the whole genome. Many computational tools have been designed to find the peaks of the signals corresponding to protein binding sites. In this paper, three computational methods, ChIP-seq processing pipeline (spp), PeakSeq and CisGenome, used in ChIP-seq data analysis are reviewed. There is also a comparison of how they agree and disagree on finding peaks using the publically available Signal Transducers and Activators of Transcription protein 1 (STAT1) and RNA polymerase II (PolII) datasets with corresponding negative controls.
染色质免疫沉淀结合大规模平行测序(ChIP-seq)是一种在全基因组范围内检测蛋白质-DNA 相互作用的有效实验策略。许多计算工具被设计用来寻找对应蛋白质结合位点信号的峰。本文综述了 ChIP-seq 数据分析中使用的三种计算方法,即 ChIP-seq 处理管道(spp)、PeakSeq 和 CisGenome。还比较了它们在使用公共转录信号转导子和激活子 1(STAT1)和 RNA 聚合酶 II(PolII)数据集及其相应阴性对照来寻找峰时的一致性和分歧。