Department of Physiology, University of Manitoba, Winnipeg, Manitoba, Canada.
PLoS One. 2011 Jan 31;6(1):e16523. doi: 10.1371/journal.pone.0016523.
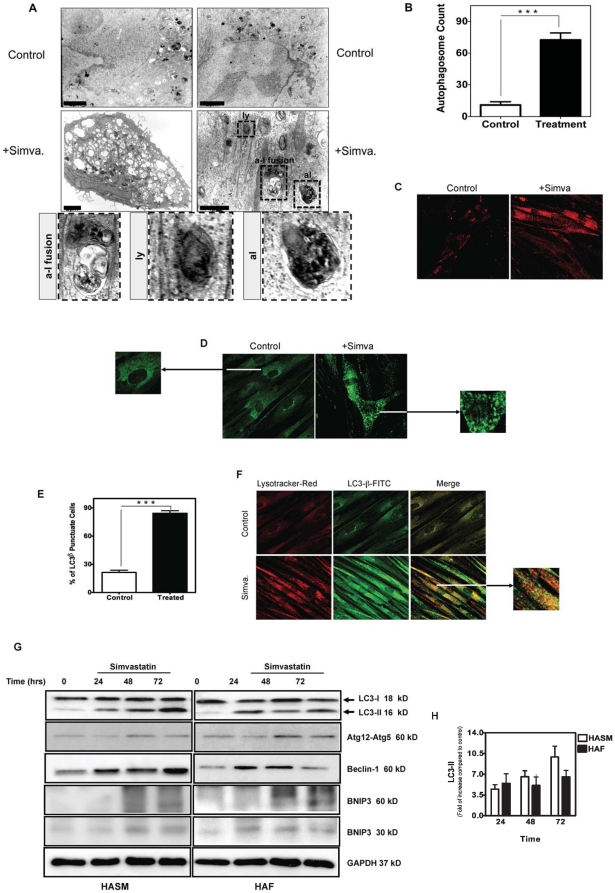
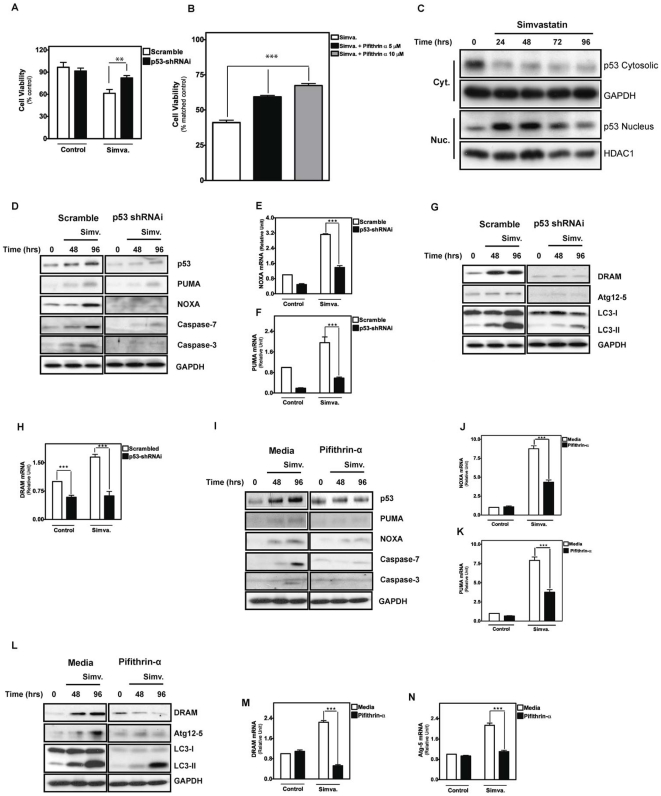
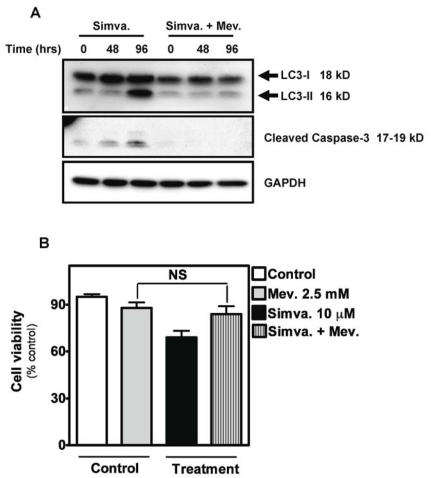
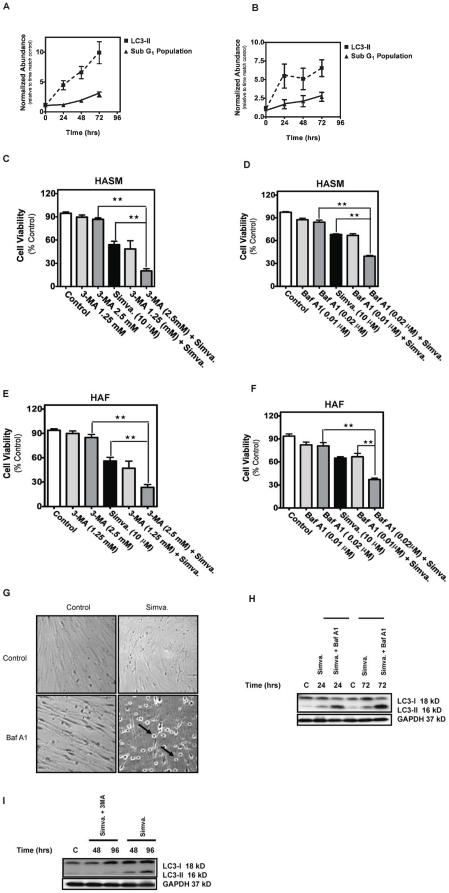
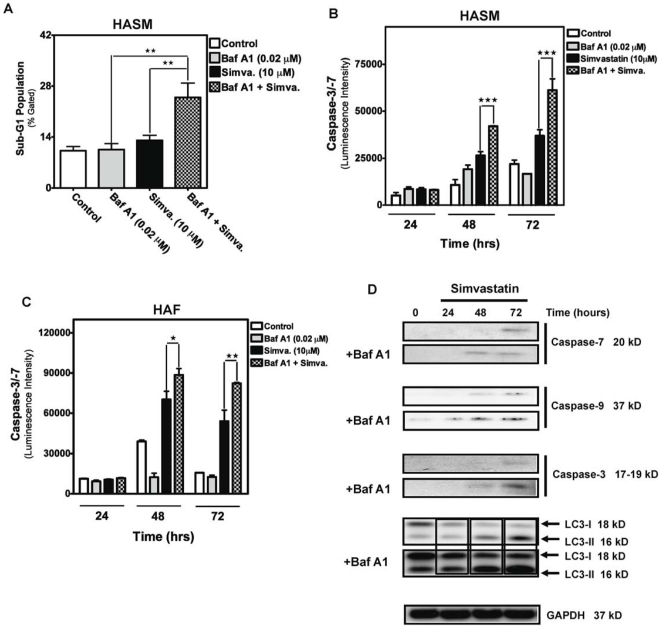
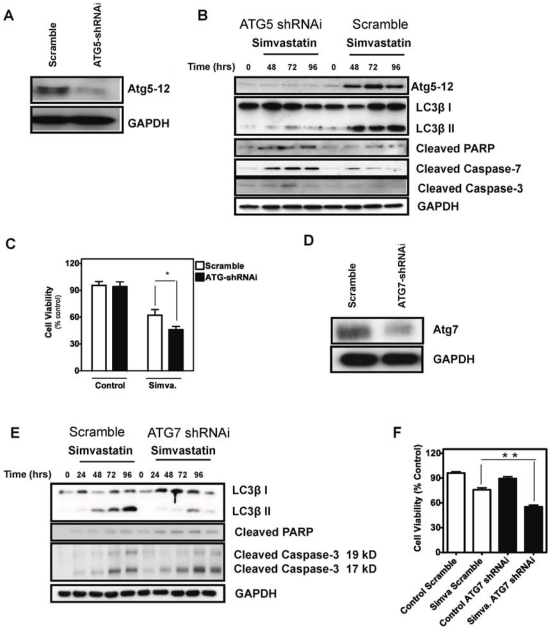
Statins inhibit the proximal steps of cholesterol biosynthesis, and are linked to health benefits in various conditions, including cancer and lung disease. We have previously investigated apoptotic pathways triggered by statins in airway mesenchymal cells, and identified reduced prenylation of small GTPases as a primary effector mechanism leading to p53-mediated cell death. Here, we extend our studies of statin-induced cell death by assessing endpoints of both apoptosis and autophagy, and investigating their interplay and coincident regulation. Using primary cultured human airway smooth muscle (HASM) and human airway fibroblasts (HAF), autophagy, and autophagosome formation and flux were assessed by transmission electron microscopy, cytochemistry (lysosome number and co-localization with LC3) and immunoblotting (LC3 lipidation and Atg12-5 complex formation). Chemical inhibition of autophagy increased simvastatin-induced caspase activation and cell death. Similarly, Atg5 silencing with shRNA, thus preventing Atg5-12 complex formation, increased pro-apoptotic effects of simvastatin. Simvastatin concomitantly increased p53-dependent expression of p53 up-regulated modulator of apoptosis (PUMA), NOXA, and damage-regulated autophagy modulator (DRAM). Notably both mevalonate cascade inhibition-induced autophagy and apoptosis were p53 dependent: simvastatin increased nuclear p53 accumulation, and both cyclic pifithrin-α and p53 shRNAi partially inhibited NOXA, PUMA expression and caspase-3/7 cleavage (apoptosis) and DRAM expression, Atg5-12 complex formation, LC3 lipidation, and autophagosome formation (autophagy). Furthermore, the autophagy response is induced rapidly, significantly delaying apoptosis, suggesting the existence of a temporally coordinated p53 regulation network. These findings are relevant for the development of statin-based therapeutic approaches in obstructive airway disease.
他汀类药物抑制胆固醇生物合成的近端步骤,并与各种情况下的健康益处相关,包括癌症和肺部疾病。我们之前研究了他汀类药物在气道间质细胞中引发的凋亡途径,并确定了小 GTPase 的预修饰减少是导致 p53 介导的细胞死亡的主要效应机制。在这里,我们通过评估凋亡和自噬的终点来扩展我们对他汀类药物诱导的细胞死亡的研究,并研究它们的相互作用和并发调节。使用原代培养的人气道平滑肌 (HASM) 和人气道成纤维细胞 (HAF),通过透射电子显微镜、细胞化学(溶酶体数量和 LC3 的共定位)和免疫印迹(LC3 脂质化和 Atg12-5 复合物形成)评估自噬和自噬体形成和通量。化学抑制自噬增加了辛伐他汀诱导的半胱天冬酶激活和细胞死亡。同样,用 shRNA 沉默 Atg5,从而阻止 Atg5-12 复合物的形成,增加了辛伐他汀的促凋亡作用。辛伐他汀同时增加了 p53 依赖性凋亡上调调节剂 (PUMA)、NOXA 和损伤调节自噬调节剂 (DRAM) 的表达。值得注意的是,甲羟戊酸途径抑制诱导的自噬和凋亡均依赖于 p53:辛伐他汀增加核 p53 积累,环匹法他汀-α和 p53 shRNAi 均可部分抑制 NOXA、PUMA 表达和 caspase-3/7 切割(凋亡)和 DRAM 表达、Atg5-12 复合物形成、LC3 脂质化和自噬体形成(自噬)。此外,自噬反应迅速诱导,显著延迟凋亡,表明存在一个时间协调的 p53 调节网络。这些发现与开发他汀类药物治疗阻塞性气道疾病的方法有关。