Naruo Yoshimi, Nagashima Takeshi, Ushikoshi-Nakayama Ryoko, Saeki Yuko, Nakakuki Takashi, Naka Takashi, Tanaka Hiroshi, Tsai Shih-Feng, Okada-Hatakeyama Mariko
Laboratory for Cellular Systems Modeling, RIKEN Research Center for Allergy and Immunology (RCAI), 1-7-22 Suehiro-cho, Tsurumi-ku, Yokohama, Kanagawa 230-0045, Japan.
BMC Syst Biol. 2011 Feb 18;5:29. doi: 10.1186/1752-0509-5-29.
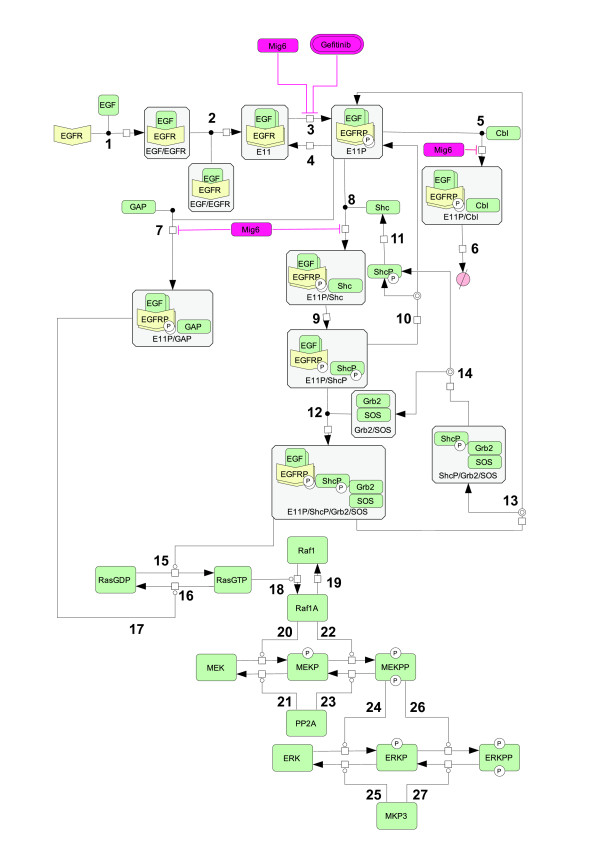
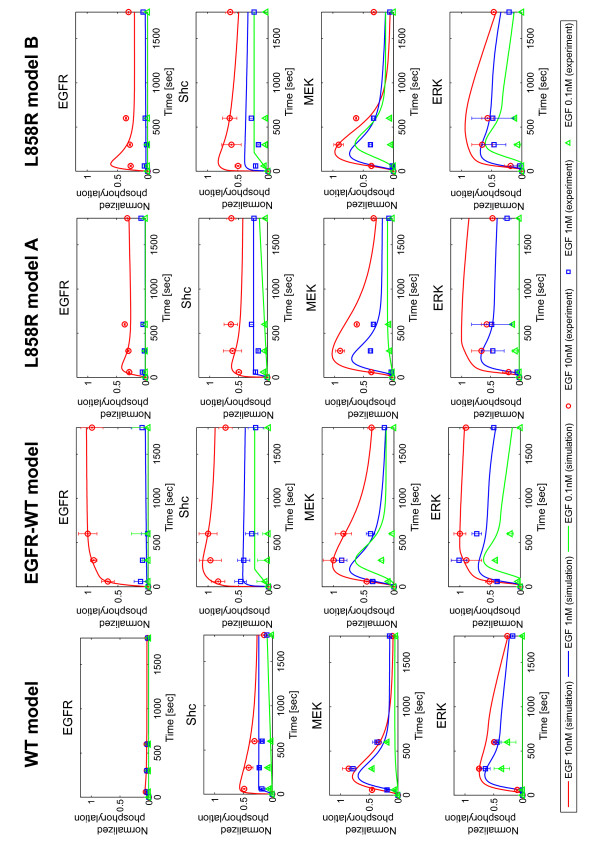
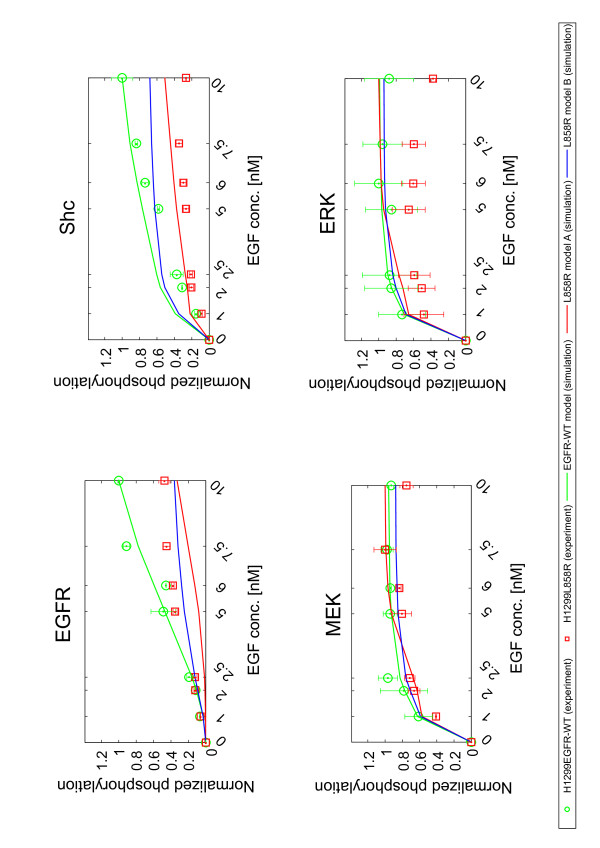
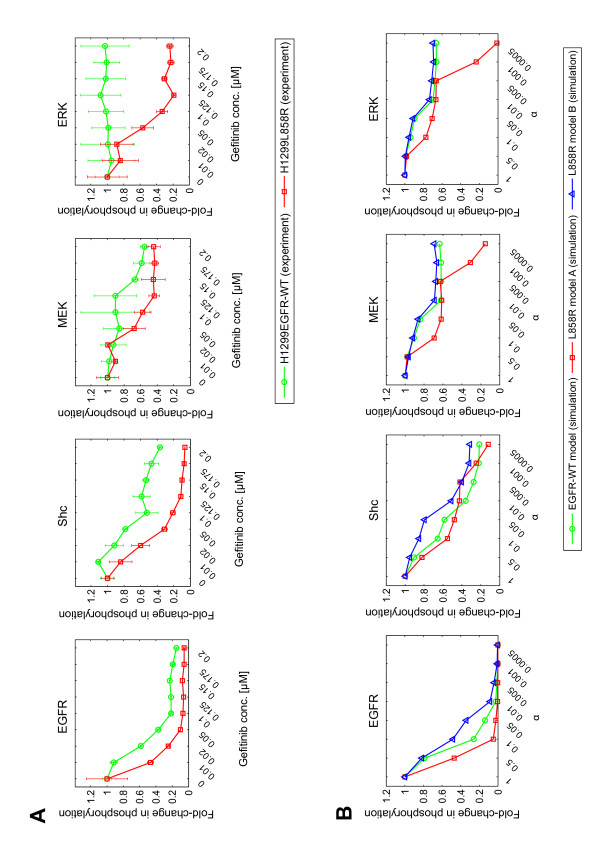
Epidermal growth factor receptor (EGFR) signaling plays an important role in the regulation of cell proliferation, survival, metastasis, and invasion in various tumors. Earlier studies showed that the EGFR is frequently overexpressed in non-small-cell lung cancer (NSCLC) and EGFR mutations at specific amino acid residues in the kinase domain induce altered responsiveness to gefitinib, a small molecule EGFR tyrosine kinase inhibitor. However, the mechanism underlying the drug response modulated by EGFR mutation is still largely unknown. To elucidate drug response in EGFR signal transduction pathway in which complex dynamics of multiple molecules involved, a systematic approach is necessary. In this paper, we performed experimental and computational analyses to clarify the underlying mechanism of EGFR signaling and cell-specific gefitinib responsiveness in three H1299-derived NSCLC cell lines; H1299 wild type (H1299WT), H1299 with an overexpressed wild type EGFR (H1299EGFR-WT), and H1299 with an overexpressed mutant EGFR L858R (H1299L858R; gefitinib sensitive mutant).
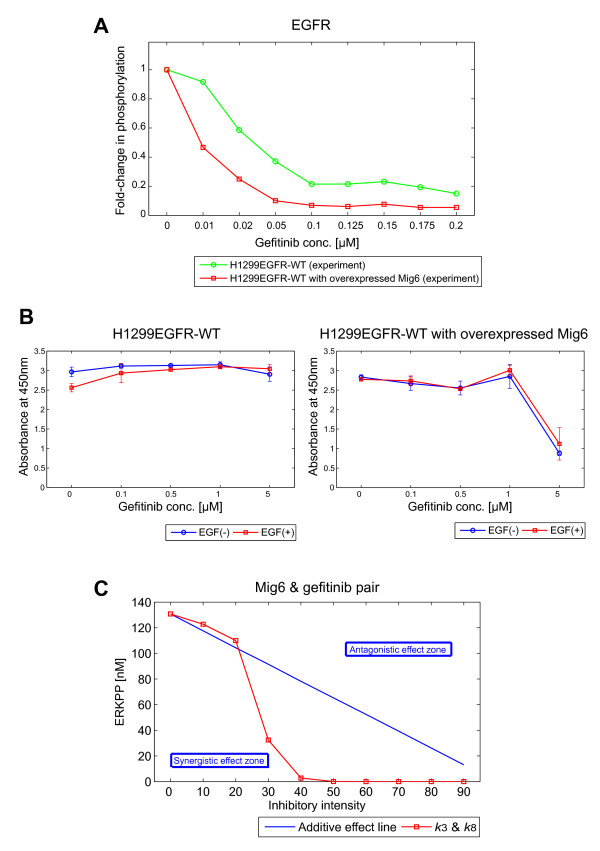
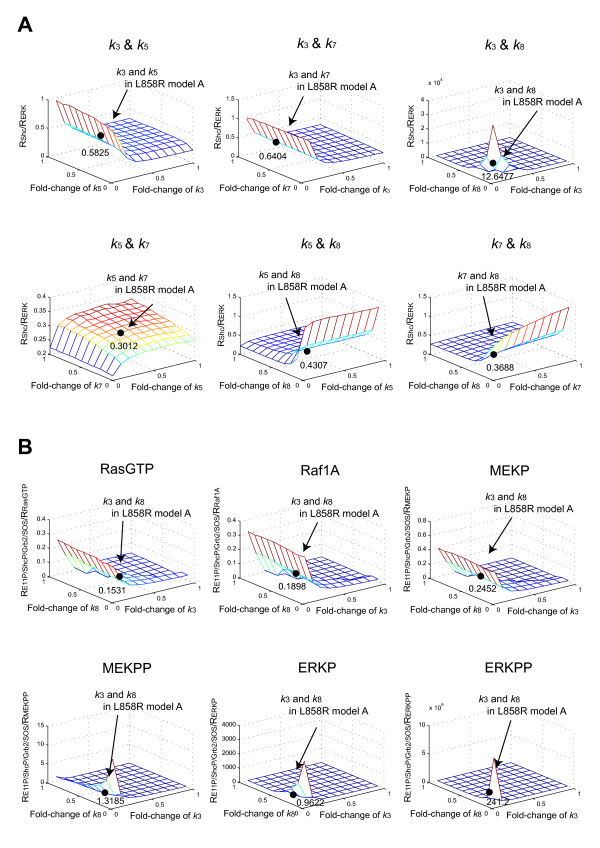
We predicted and experimentally verified that Mig6, which is a known negative regulator of EGFR and specifically expressed in H1299L858R cells, synergized with gefitinib to suppress cellular growth. Computational analyses indicated that this inhibitory effect is amplified at the phosphorylation/dephosphorylation steps of MEK and ERK.
Thus, we showed that L858R receptor mutation in combination with expression of its negative regulator, Mig6, alters signaling outcomes and results in variable drug sensitivity.
表皮生长因子受体(EGFR)信号传导在多种肿瘤的细胞增殖、存活、转移和侵袭调节中发挥重要作用。早期研究表明,EGFR在非小细胞肺癌(NSCLC)中经常过度表达,激酶结构域中特定氨基酸残基的EGFR突变会导致对小分子EGFR酪氨酸激酶抑制剂吉非替尼的反应性改变。然而,EGFR突变调节药物反应的潜在机制仍 largely未知。为了阐明涉及多个分子复杂动力学的EGFR信号转导途径中的药物反应,需要一种系统的方法。在本文中,我们进行了实验和计算分析,以阐明三种H1299衍生的NSCLC细胞系中EGFR信号传导和细胞特异性吉非替尼反应性的潜在机制;H1299野生型(H1299WT)、过表达野生型EGFR的H1299(H1299EGFR-WT)和过表达突变型EGFR L858R的H1299(H1299L858R;吉非替尼敏感突变体)。
我们预测并通过实验验证,Mig6是一种已知的EGFR负调节剂,在H1299L858R细胞中特异性表达,它与吉非替尼协同抑制细胞生长。计算分析表明,这种抑制作用在MEK和ERK的磷酸化/去磷酸化步骤中被放大。
因此,我们表明L858R受体突变与其负调节剂Mig6的表达相结合,改变了信号传导结果并导致可变的药物敏感性。