Department of Biosciences and Informatics, Keio University, 3-14-1 Hiyoshi, Kohoku-ku, Yokohama, Kanagawa 223-8522, Japan.
BMC Bioinformatics. 2011 Feb 15;12 Suppl 1(Suppl 1):S48. doi: 10.1186/1471-2105-12-S1-S48.
Clustering of unannotated transcripts is an important task to identify novel families of noncoding RNAs (ncRNAs). Several hierarchical clustering methods have been developed using similarity measures based on the scores of structural alignment. However, the high computational cost of exact structural alignment requires these methods to employ approximate algorithms. Such heuristics degrade the quality of clustering results, especially when the similarity among family members is not detectable at the primary sequence level.
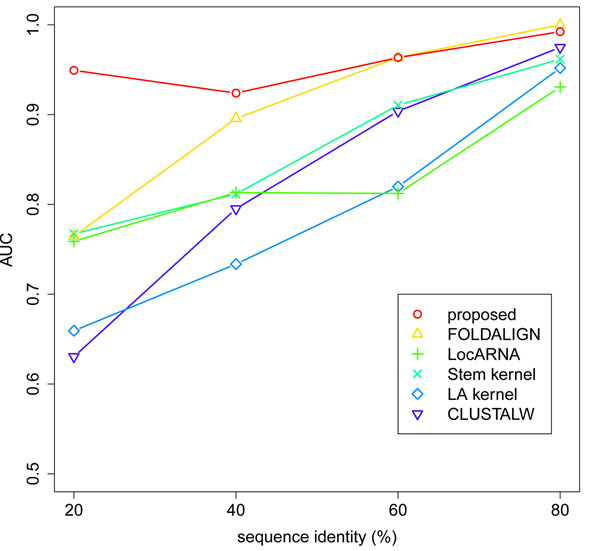
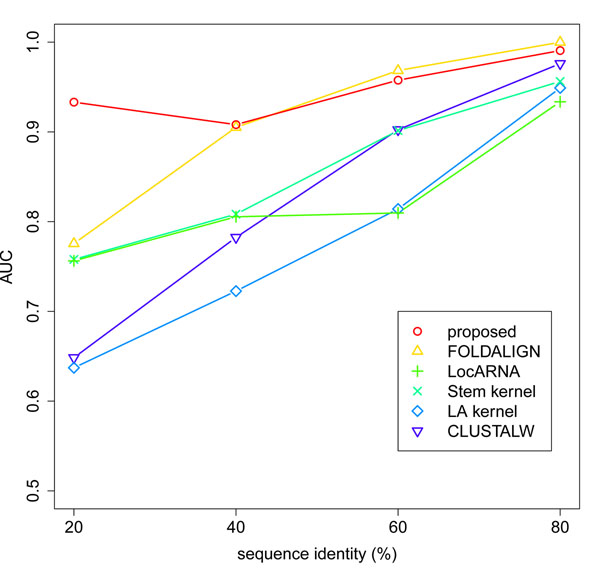
We describe a new similarity measure for the hierarchical clustering of ncRNAs. The idea is that the reliability of approximate algorithms can be improved by utilizing the information of suboptimal solutions in their dynamic programming frameworks. We approximate structural alignment in a more simplified manner than the existing methods. Instead, our method utilizes all possible sequence alignments and all possible secondary structures, whereas the existing methods only use one optimal sequence alignment and one optimal secondary structure. We demonstrate that this strategy can achieve the best balance between the computational cost and the quality of the clustering. In particular, our method can keep its high performance even when the sequence identity of family members is less than 60%.
Our method enables fast and accurate clustering of ncRNAs. The software is available for download at http://bpla-kernel.dna.bio.keio.ac.jp/clustering/.
对未注释的转录本进行聚类是识别新的非编码 RNA(ncRNA)家族的重要任务。已经开发了几种基于结构比对得分的相似性度量的层次聚类方法。然而,精确结构比对的计算成本很高,这要求这些方法采用近似算法。这种启发式方法会降低聚类结果的质量,尤其是在家族成员之间的相似性在一级序列水平上无法检测到时。
我们描述了一种用于 ncRNA 层次聚类的新相似性度量方法。其思想是,通过在动态规划框架中利用次优解的信息,可以提高近似算法的可靠性。我们以比现有方法更简化的方式进行结构比对的近似处理。相反,我们的方法利用了所有可能的序列比对和所有可能的二级结构,而现有方法仅使用一个最优的序列比对和一个最优的二级结构。我们证明了这种策略可以在计算成本和聚类质量之间达到最佳平衡。特别是,即使家族成员的序列同一性小于 60%,我们的方法也能保持高性能。
我们的方法能够快速准确地对 ncRNA 进行聚类。该软件可在 http://bpla-kernel.dna.bio.keio.ac.jp/clustering/ 下载。