Will Sebastian, Otto Christina, Miladi Milad, Möhl Mathias, Backofen Rolf
Bioinformatics, Department of Computer Science, University of Freiburg, Freiburg, Germany, Bioinformatics, Department of Computer Science, University of Leipzig, Leipzig, Germany.
Bioinformatics, Department of Computer Science, University of Freiburg, Freiburg, Germany.
Bioinformatics. 2015 Aug 1;31(15):2489-96. doi: 10.1093/bioinformatics/btv185. Epub 2015 Apr 2.
RNA-Seq experiments have revealed a multitude of novel ncRNAs. The gold standard for their analysis based on simultaneous alignment and folding suffers from extreme time complexity of [Formula: see text]. Subsequently, numerous faster 'Sankoff-style' approaches have been suggested. Commonly, the performance of such methods relies on sequence-based heuristics that restrict the search space to optimal or near-optimal sequence alignments; however, the accuracy of sequence-based methods breaks down for RNAs with sequence identities below 60%. Alignment approaches like LocARNA that do not require sequence-based heuristics, have been limited to high complexity ([Formula: see text] quartic time).
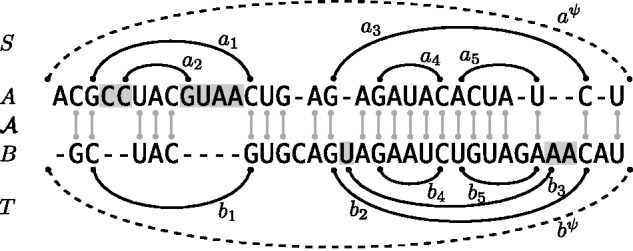
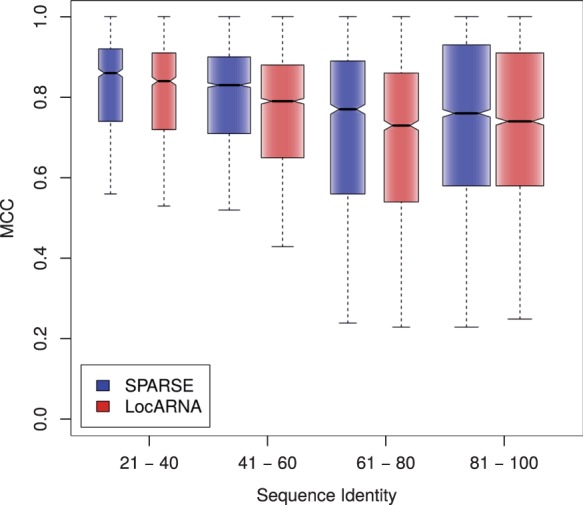
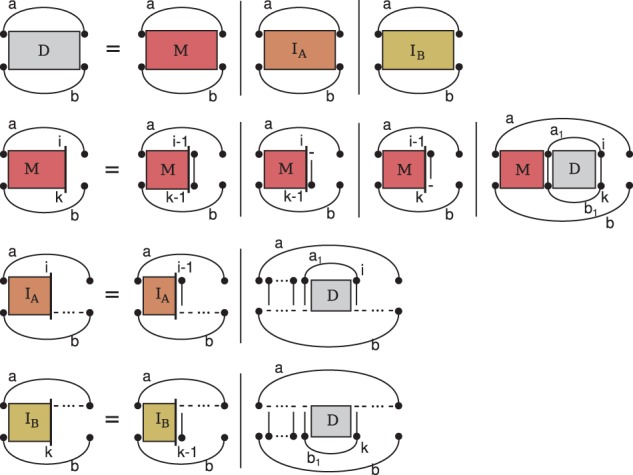
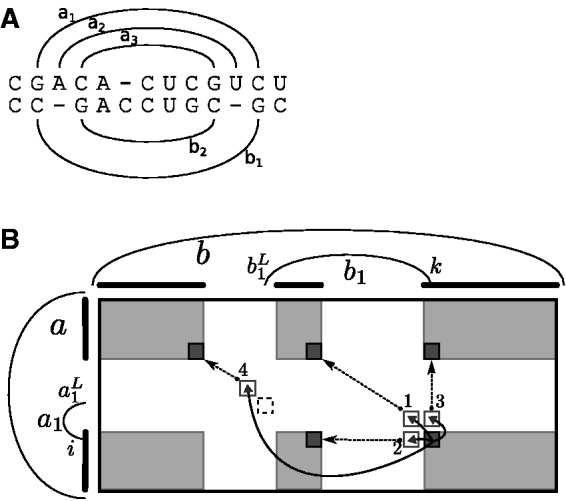
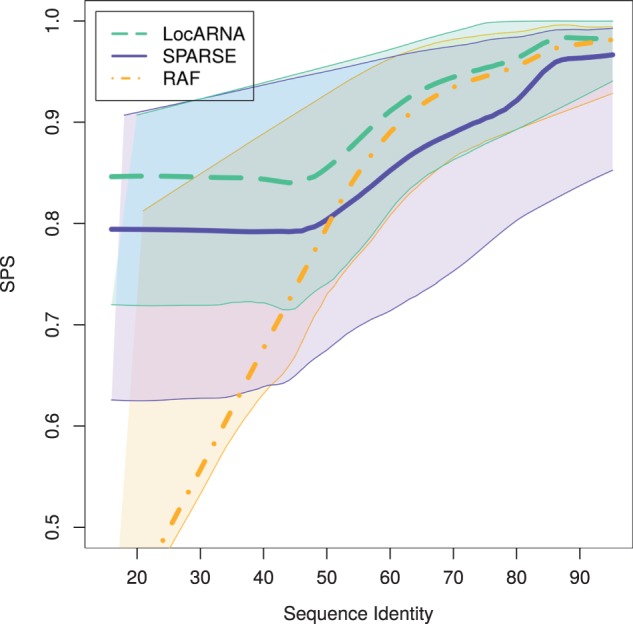
Breaking this barrier, we introduce the novel Sankoff-style algorithm 'sparsified prediction and alignment of RNAs based on their structure ensembles (SPARSE)', which runs in quadratic time without sequence-based heuristics. To achieve this low complexity, on par with sequence alignment algorithms, SPARSE features strong sparsification based on structural properties of the RNA ensembles. Following PMcomp, SPARSE gains further speed-up from lightweight energy computation. Although all existing lightweight Sankoff-style methods restrict Sankoff's original model by disallowing loop deletions and insertions, SPARSE transfers the Sankoff algorithm to the lightweight energy model completely for the first time. Compared with LocARNA, SPARSE achieves similar alignment and better folding quality in significantly less time (speedup: 3.7). At similar run-time, it aligns low sequence identity instances substantially more accurate than RAF, which uses sequence-based heuristics.
RNA测序实验揭示了大量新型非编码RNA。基于同时比对和折叠对其进行分析的金标准存在[公式:见原文]的极端时间复杂度问题。随后,人们提出了许多更快的“桑科夫风格”方法。通常,这些方法的性能依赖于基于序列的启发式算法,将搜索空间限制在最优或接近最优的序列比对上;然而,对于序列同一性低于60%的RNA,基于序列的方法的准确性会下降。像LocARNA这样不需要基于序列启发式算法的比对方法,其时间复杂度被限制在高复杂度([公式:见原文]四次方时间)。
我们打破了这一障碍,引入了新颖的桑科夫风格算法“基于RNA结构集合的稀疏预测与比对(SPARSE)”,该算法在不使用基于序列启发式算法的情况下以二次时间运行。为了实现这种低复杂度,与序列比对算法相当,SPARSE基于RNA集合的结构特性进行了强大的稀疏化处理。遵循PMcomp,SPARSE通过轻量级能量计算进一步加速。尽管所有现有的轻量级桑科夫风格方法都通过禁止环的删除和插入来限制桑科夫的原始模型,但SPARSE首次将桑科夫算法完全转移到轻量级能量模型中。与LocARNA相比,SPARSE在显著更短的时间内(加速比:3.7)实现了相似的比对和更好的折叠质量。在相似的运行时间下,它比对低序列同一性实例的准确性比使用基于序列启发式算法的RAF高得多。