Bioinformatics Laboratory, Max Planck Institute of Molecular Cell Biology and Genetics, Dresden, Saxony, Germany.
PLoS One. 2011 Mar 10;6(3):e17568. doi: 10.1371/journal.pone.0017568.
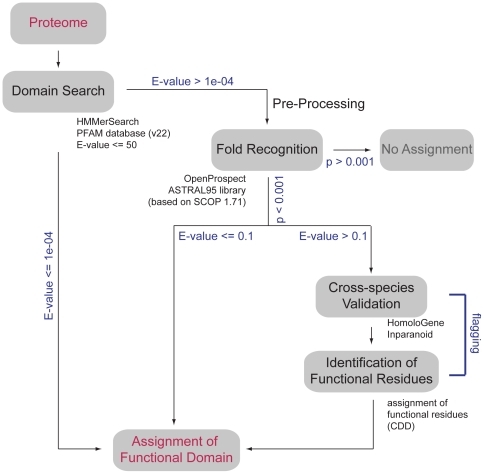
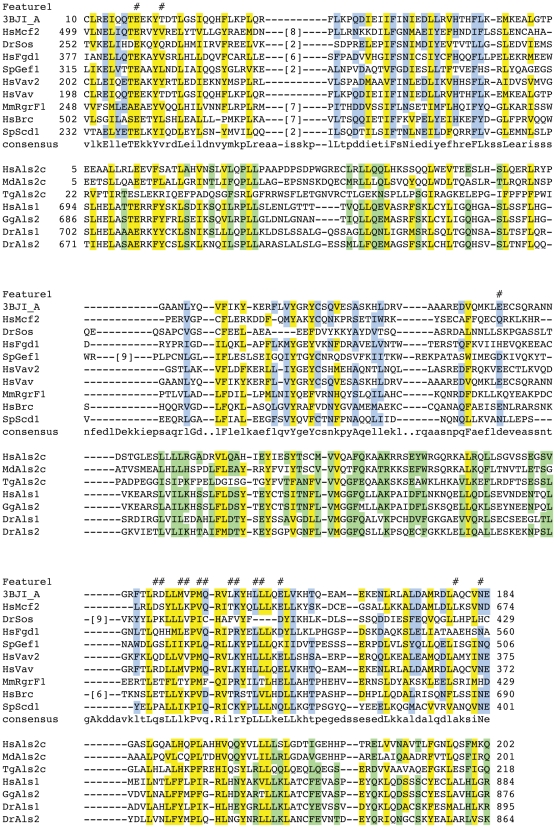
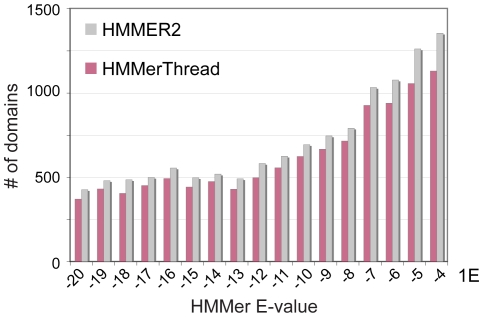
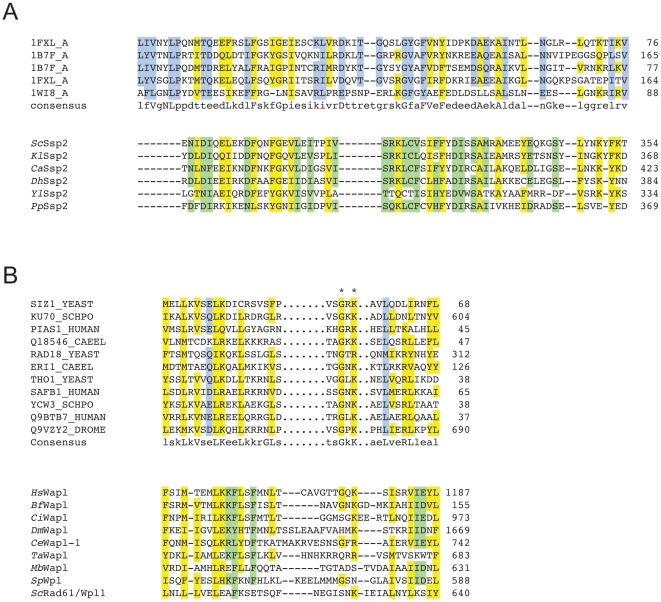
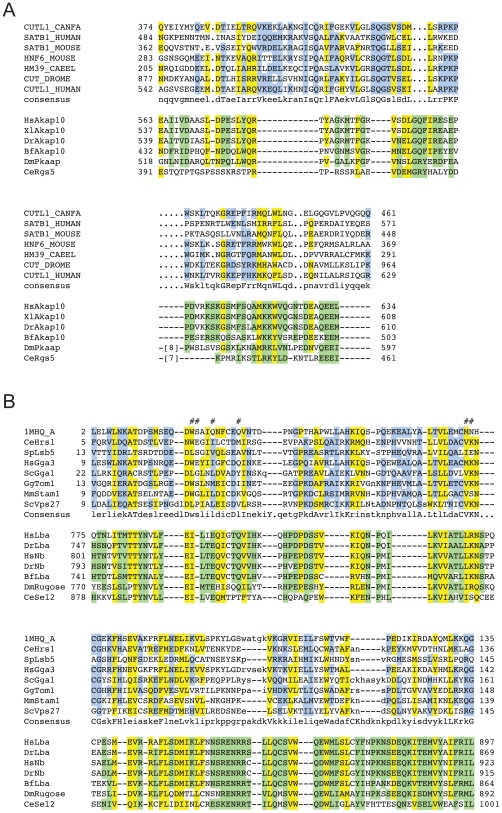
Conserved domains in proteins are one of the major sources of functional information for experimental design and genome-level annotation. Though search tools for conserved domain databases such as Hidden Markov Models (HMMs) are sensitive in detecting conserved domains in proteins when they share sufficient sequence similarity, they tend to miss more divergent family members, as they lack a reliable statistical framework for the detection of low sequence similarity. We have developed a greatly improved HMMerThread algorithm that can detect remotely conserved domains in highly divergent sequences. HMMerThread combines relaxed conserved domain searches with fold recognition to eliminate false positive, sequence-based identifications. With an accuracy of 90%, our software is able to automatically predict highly divergent members of conserved domain families with an associated 3-dimensional structure. We give additional confidence to our predictions by validation across species. We have run HMMerThread searches on eight proteomes including human and present a rich resource of remotely conserved domains, which adds significantly to the functional annotation of entire proteomes. We find ∼4500 cross-species validated, remotely conserved domain predictions in the human proteome alone. As an example, we find a DNA-binding domain in the C-terminal part of the A-kinase anchor protein 10 (AKAP10), a PKA adaptor that has been implicated in cardiac arrhythmias and premature cardiac death, which upon stress likely translocates from mitochondria to the nucleus/nucleolus. Based on our prediction, we propose that with this HLH-domain, AKAP10 is involved in the transcriptional control of stress response. Further remotely conserved domains we discuss are examples from areas such as sporulation, chromosome segregation and signalling during immune response. The HMMerThread algorithm is able to automatically detect the presence of remotely conserved domains in proteins based on weak sequence similarity. Our predictions open up new avenues for biological and medical studies. Genome-wide HMMerThread domains are available at http://vm1-hmmerthread.age.mpg.de.
蛋白质中的保守结构域是实验设计和全基因组注释的主要功能信息来源之一。尽管像隐马尔可夫模型(HMM)这样的保守结构域数据库搜索工具在蛋白质具有足够的序列相似性时,能够敏感地检测到保守结构域,但它们往往会错过更多具有差异的家族成员,因为它们缺乏可靠的统计框架来检测低序列相似性。我们开发了一种大大改进的 HMMerThread 算法,该算法可以在高度差异的序列中检测到远程保守结构域。HMMerThread 将宽松的保守结构域搜索与折叠识别相结合,以消除假阳性的基于序列的鉴定。我们的软件准确性达到 90%,能够自动预测具有相关三维结构的保守结构域家族的高度差异成员。通过跨物种验证,我们为预测提供了额外的置信度。我们在包括人类在内的八个蛋白质组上运行了 HMMerThread 搜索,并提供了丰富的远程保守结构域资源,这极大地增加了整个蛋白质组的功能注释。我们仅在人类蛋白质组中就发现了约 4500 个跨物种验证的、远程保守结构域预测。例如,我们在 A-激酶锚定蛋白 10(AKAP10)的 C 端部分发现了一个 DNA 结合结构域,AKAP10 是一种 PKA 接头,与心脏心律失常和心脏性猝死有关,在应激下可能从线粒体易位到核/核仁。基于我们的预测,我们提出 AKAP10 通过这个 HLH 结构域参与应激反应的转录控制。我们还讨论了其他一些远程保守结构域,例如孢子形成、染色体分离和免疫反应期间的信号转导。HMMerThread 算法能够基于弱序列相似性自动检测蛋白质中远程保守结构域的存在。我们的预测为生物学和医学研究开辟了新的途径。全基因组 HMMerThread 结构域可在 http://vm1-hmmerthread.age.mpg.de 获得。