RIKEN Biomass Engineering Program, Yokohama 230-0045, Japan.
Plant Cell Physiol. 2011 May;52(5):785-803. doi: 10.1093/pcp/pcr035. Epub 2011 Mar 24.
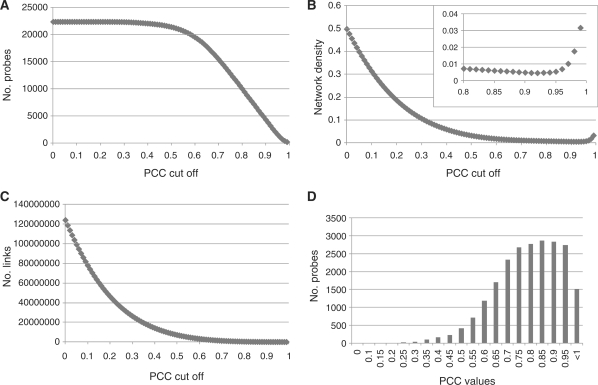
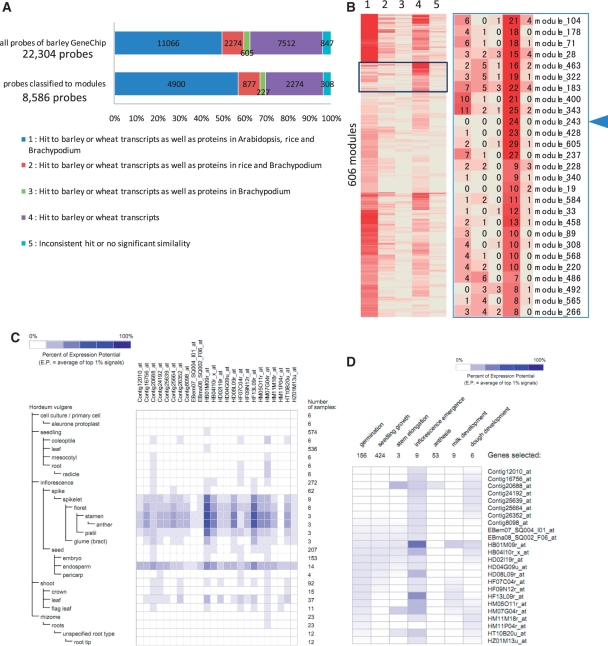
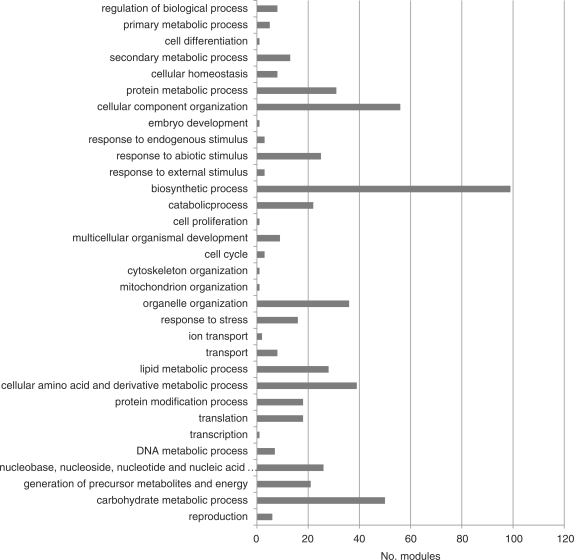
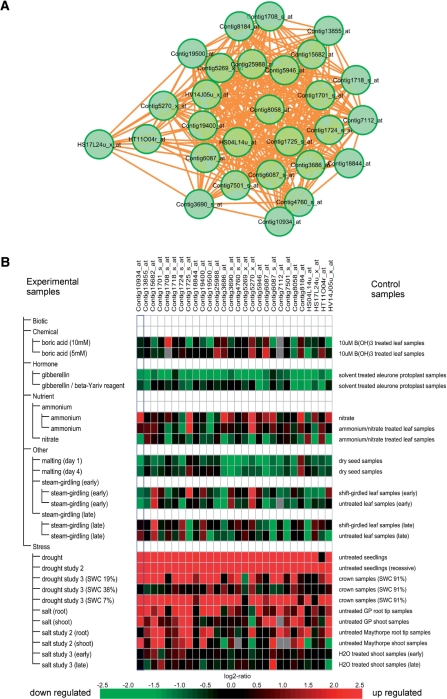
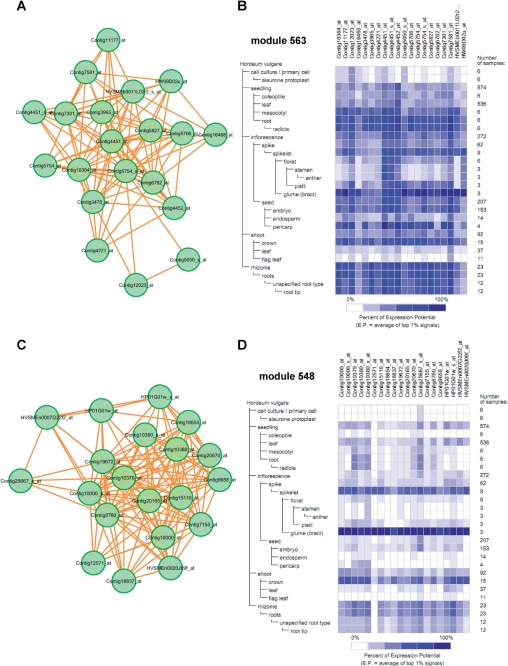
Accumulated transcriptome data can be used to investigate regulatory networks of genes involved in various biological systems. Co-expression analysis data sets generated from comprehensively collected transcriptome data sets now represent efficient resources that are capable of facilitating the discovery of genes with closely correlated expression patterns. In order to construct a co-expression network for barley, we analyzed 45 publicly available experimental series, which are composed of 1,347 sets of GeneChip data for barley. On the basis of a gene-to-gene weighted correlation coefficient, we constructed a global barley co-expression network and classified it into clusters of subnetwork modules. The resulting clusters are candidates for functional regulatory modules in the barley transcriptome. To annotate each of the modules, we performed comparative annotation using genes in Arabidopsis and Brachypodium distachyon. On the basis of a comparative analysis between barley and two model species, we investigated functional properties from the representative distributions of the gene ontology (GO) terms. Modules putatively involved in drought stress response and cellulose biogenesis have been identified. These modules are discussed to demonstrate the effectiveness of the co-expression analysis. Furthermore, we applied the data set of co-expressed genes coupled with comparative analysis in attempts to discover potentially Triticeae-specific network modules. These results demonstrate that analysis of the co-expression network of the barley transcriptome together with comparative analysis should promote the process of gene discovery in barley. Furthermore, the insights obtained should be transferable to investigations of Triticeae plants. The associated data set generated in this analysis is publicly accessible at http://coexpression.psc.riken.jp/barley/.
积累的转录组数据可用于研究参与各种生物系统的基因的调控网络。现在,从全面收集的转录组数据集生成的共表达分析数据集代表了有效的资源,能够促进发现具有密切相关表达模式的基因。为了构建大麦的共表达网络,我们分析了 45 个公开可用的实验系列,这些系列由 1347 组大麦基因芯片数据组成。基于基因间加权相关系数,我们构建了一个全局大麦共表达网络,并将其分类为子网模块的簇。所得聚类是大麦转录组中功能调节模块的候选者。为了注释每个模块,我们使用拟南芥和短柄草中的基因进行了比较注释。基于大麦与两种模式物种之间的比较分析,我们从基因本体 (GO) 术语的代表性分布中研究了功能特性。鉴定了可能参与干旱胁迫反应和纤维素生物发生的模块。讨论了这些模块,以证明共表达分析的有效性。此外,我们应用了共表达基因数据集和比较分析,试图发现潜在的小麦族特异性网络模块。这些结果表明,分析大麦转录组的共表达网络并结合比较分析应促进大麦基因发现的过程。此外,获得的见解应该可以转移到对小麦族植物的研究。在此分析中生成的相关数据集可在 http://coexpression.psc.riken.jp/barley/ 上公开获取。