Hung Ling-Hong, Guerquin Michal, Samudrala Ram
Department of Microbiology, University of Washington, Seattle WA USA.
BMC Res Notes. 2011 Apr 1;4:97. doi: 10.1186/1756-0500-4-97.
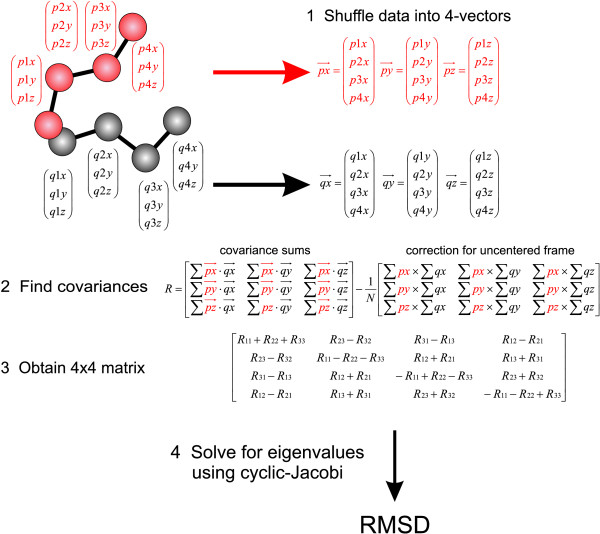
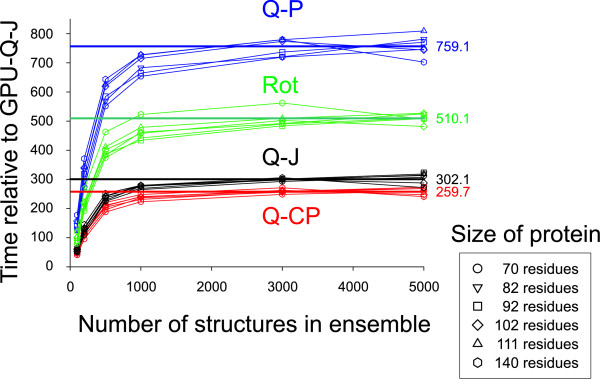
Calculation of the root mean square deviation (RMSD) between the atomic coordinates of two optimally superposed structures is a basic component of structural comparison techniques. We describe a quaternion based method, GPU-Q-J, that is stable with single precision calculations and suitable for graphics processor units (GPUs). The application was implemented on an ATI 4770 graphics card in C/C++ and Brook+ in Linux where it was 260 to 760 times faster than existing unoptimized CPU methods. Source code is available from the Compbio website http://software.compbio.washington.edu/misc/downloads/st_gpu_fit/ or from the author LHH.
The Nutritious Rice for the World Project (NRW) on World Community Grid predicted de novo, the structures of over 62,000 small proteins and protein domains returning a total of 10 billion candidate structures. Clustering ensembles of structures on this scale requires calculation of large similarity matrices consisting of RMSDs between each pair of structures in the set. As a real-world test, we calculated the matrices for 6 different ensembles from NRW. The GPU method was 260 times faster that the fastest existing CPU based method and over 500 times faster than the method that had been previously used.
GPU-Q-J is a significant advance over previous CPU methods. It relieves a major bottleneck in the clustering of large numbers of structures for NRW. It also has applications in structure comparison methods that involve multiple superposition and RMSD determination steps, particularly when such methods are applied on a proteome and genome wide scale.
计算两个最优叠加结构的原子坐标之间的均方根偏差(RMSD)是结构比较技术的基本组成部分。我们描述了一种基于四元数的方法GPU-Q-J,它在单精度计算中稳定且适用于图形处理器(GPU)。该应用程序是在Linux系统中使用C/C++和Brook+在ATI 4770图形卡上实现的,其速度比现有的未优化CPU方法快260到760倍。源代码可从Compbio网站http://software.compbio.washington.edu/misc/downloads/st_gpu_fit/获取,或从作者LHH处获取。
世界社区网格上的世界营养大米项目(NRW)从头预测了超过62,000个小蛋白质和蛋白质结构域的结构,共返回100亿个候选结构。对如此规模的结构集合进行聚类需要计算由集合中每对结构之间的RMSD组成的大型相似性矩阵。作为实际测试,我们计算了来自NRW的6个不同集合的矩阵。GPU方法比现有的最快CPU方法快260倍,比之前使用的方法快500倍以上。
GPU-Q-J相对于以前的CPU方法有显著进步。它缓解了NRW中大量结构聚类的一个主要瓶颈。它还在涉及多次叠加和RMSD确定步骤的结构比较方法中有应用,特别是当这些方法应用于蛋白质组和基因组范围时。