Département de Biologie Structurale et Génomique, IGBMC (Institut de Génétique et de Biologie Moléculaire et Cellulaire), CNRS/INSERM/Université de Strasbourg, Illkirch, France.
PLoS One. 2011 Mar 31;6(3):e18093. doi: 10.1371/journal.pone.0018093.
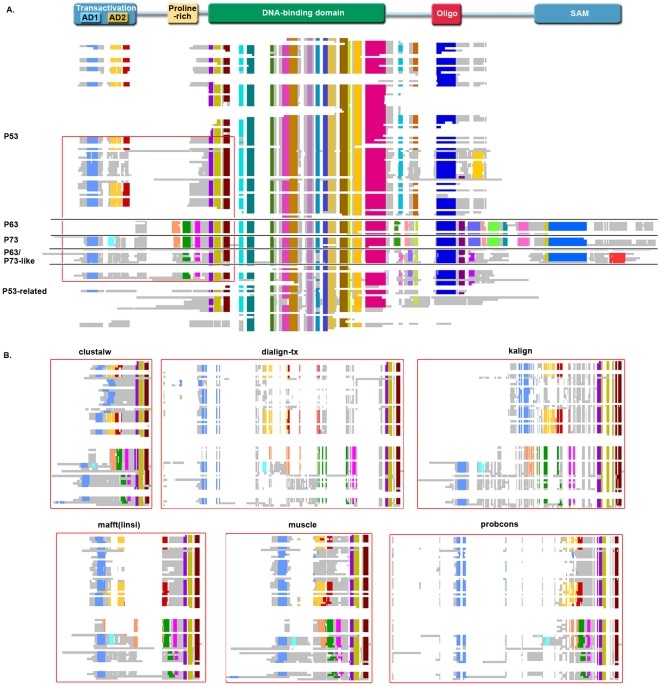
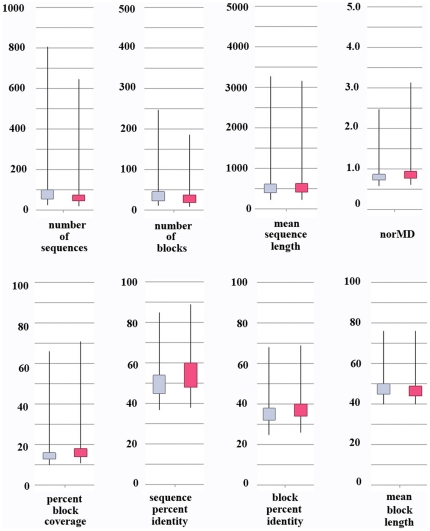
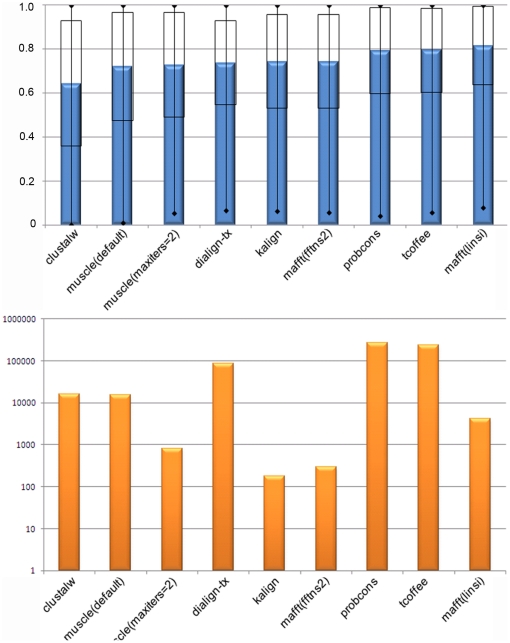
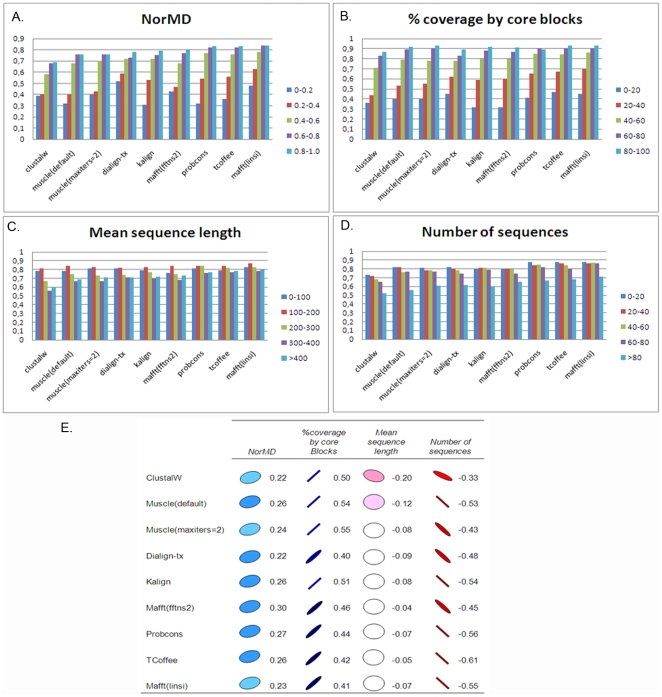
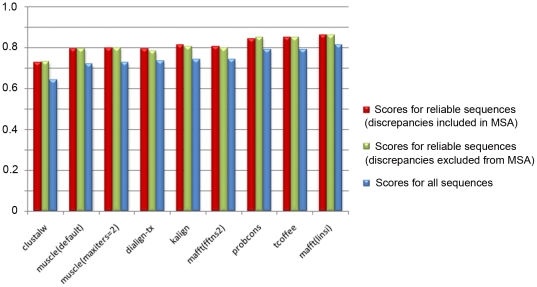
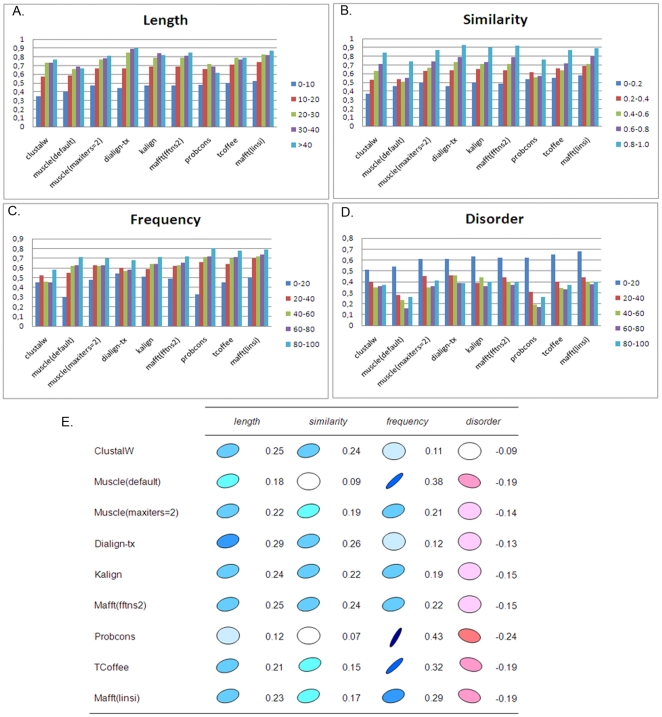
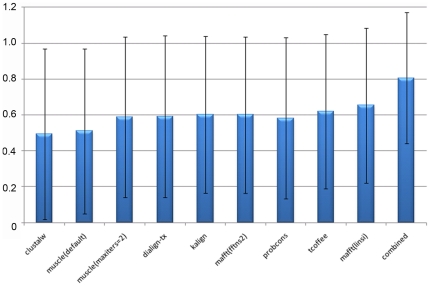
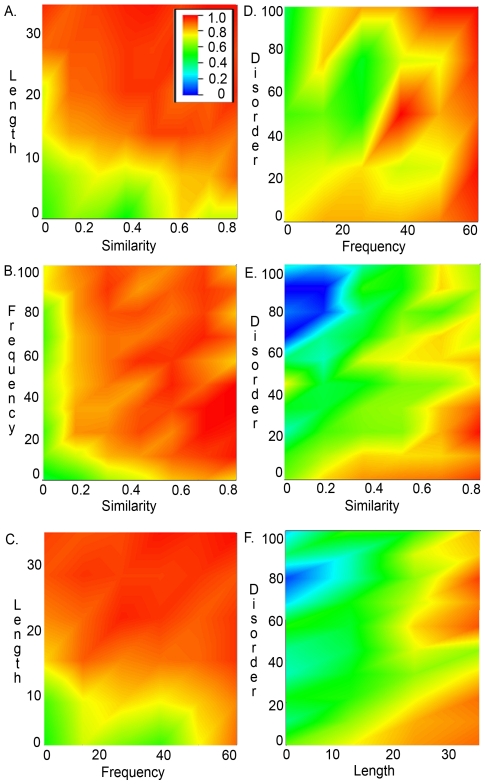
Multiple comparison or alignmentof protein sequences has become a fundamental tool in many different domains in modern molecular biology, from evolutionary studies to prediction of 2D/3D structure, molecular function and inter-molecular interactions etc. By placing the sequence in the framework of the overall family, multiple alignments can be used to identify conserved features and to highlight differences or specificities. In this paper, we describe a comprehensive evaluation of many of the most popular methods for multiple sequence alignment (MSA), based on a new benchmark test set. The benchmark is designed to represent typical problems encountered when aligning the large protein sequence sets that result from today's high throughput biotechnologies. We show that alignmentmethods have significantly progressed and can now identify most of the shared sequence features that determine the broad molecular function(s) of a protein family, even for divergent sequences. However,we have identified a number of important challenges. First, the locally conserved regions, that reflect functional specificities or that modulate a protein's function in a given cellular context,are less well aligned. Second, motifs in natively disordered regions are often misaligned. Third, the badly predicted or fragmentary protein sequences, which make up a large proportion of today's databases, lead to a significant number of alignment errors. Based on this study, we demonstrate that the existing MSA methods can be exploited in combination to improve alignment accuracy, although novel approaches will still be needed to fully explore the most difficult regions. We then propose knowledge-enabled, dynamic solutions that will hopefully pave the way to enhanced alignment construction and exploitation in future evolutionary systems biology studies.
蛋白质序列的多重比较或比对已成为现代分子生物学中许多不同领域的基本工具,从进化研究到 2D/3D 结构、分子功能和分子间相互作用的预测等。通过将序列置于整个家族的框架中,多重比对可用于识别保守特征,并突出差异或特异性。在本文中,我们基于新的基准测试集对许多最流行的多序列比对(MSA)方法进行了全面评估。该基准旨在代表在当今高通量生物技术产生的大型蛋白质序列集中进行比对时遇到的典型问题。我们表明,比对方法已经取得了显著进展,即使对于高度分化的序列,现在也能够识别决定蛋白质家族广泛分子功能的大多数共享序列特征。然而,我们也发现了一些重要的挑战。首先,局部保守区域,反映功能特异性或在特定细胞环境中调节蛋白质功能的区域,对齐效果较差。其次,天然无序区域中的模体经常被错误对齐。第三,预测不良或片段化的蛋白质序列占当今数据库的很大一部分,导致大量的比对错误。基于这项研究,我们证明可以结合使用现有的 MSA 方法来提高比对准确性,尽管仍需要新的方法来充分探索最困难的区域。然后,我们提出了基于知识的动态解决方案,希望为未来的进化系统生物学研究中的增强比对构建和利用铺平道路。