Wright Erik S
Department of Bacteriology, University of Wisconsin-Madison, Madison, WI, 53715, USA.
Wisconsin Institute for Discovery, University of Wisconsin-Madison, 330 N. Orchard St., Madison, WI, 53715, USA.
BMC Bioinformatics. 2015 Oct 6;16:322. doi: 10.1186/s12859-015-0749-z.
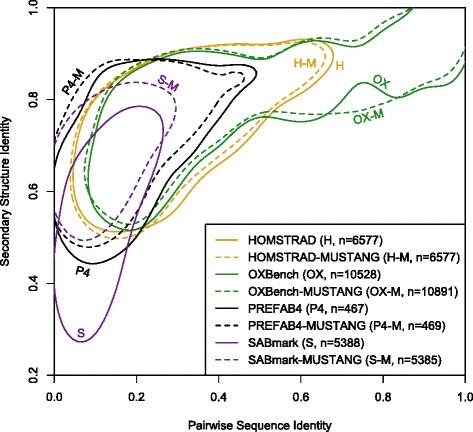
Alignment of large and diverse sequence sets is a common task in biological investigations, yet there remains considerable room for improvement in alignment quality. Multiple sequence alignment programs tend to reach maximal accuracy when aligning only a few sequences, and then diminish steadily as more sequences are added. This drop in accuracy can be partly attributed to a build-up of error and ambiguity as more sequences are aligned. Most high-throughput sequence alignment algorithms do not use contextual information under the assumption that sites are independent. This study examines the extent to which local sequence context can be exploited to improve the quality of large multiple sequence alignments.
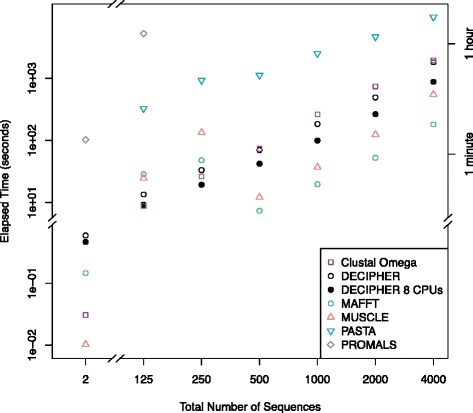
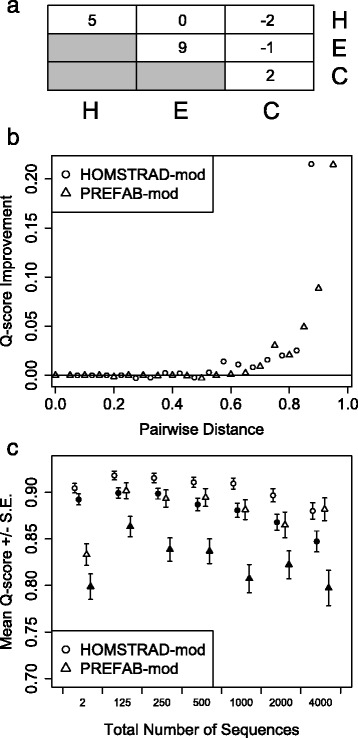
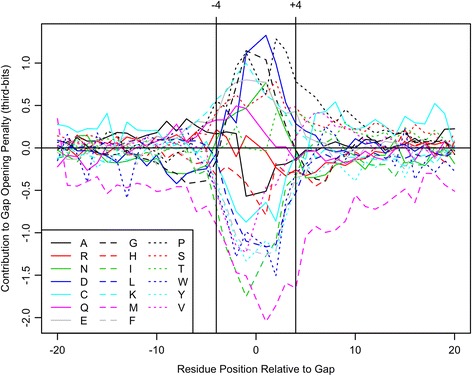
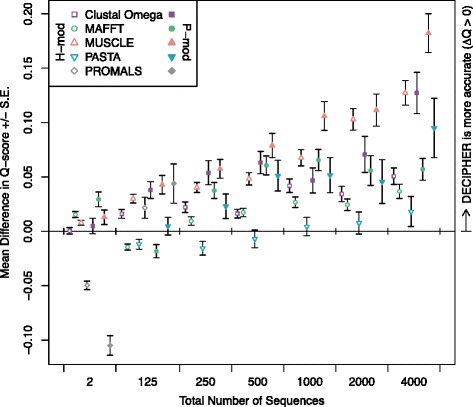
Two predictors based on local sequence context were assessed: (i) single sequence secondary structure predictions, and (ii) modulation of gap costs according to the surrounding residues. The results indicate that context-based predictors have appreciable information content that can be utilized to create more accurate alignments. Furthermore, local context becomes more informative as the number of sequences increases, enabling more accurate protein alignments of large empirical benchmarks. These discoveries became the basis for DECIPHER, a new context-aware program for sequence alignment, which outperformed other programs on large sequence sets.
Predicting secondary structure based on local sequence context is an efficient means of breaking the independence assumption in alignment. Since secondary structure is more conserved than primary sequence, it can be leveraged to improve the alignment of distantly related proteins. Moreover, secondary structure predictions increase in accuracy as more sequences are used in the prediction. This enables the scalable generation of large sequence alignments that maintain high accuracy even on diverse sequence sets. The DECIPHER R package and source code are freely available for download at DECIPHER.cee.wisc.edu and from the Bioconductor repository.
在生物学研究中,对大量不同序列集进行比对是一项常见任务,但比对质量仍有很大的提升空间。多个序列比对程序在仅比对少数序列时往往能达到最大准确度,而随着序列数量增加,准确度会稳步下降。这种准确度下降部分可归因于随着比对序列增多,错误和模糊性逐渐累积。大多数高通量序列比对算法在假设位点相互独立的情况下未使用上下文信息。本研究考察了利用局部序列上下文来提高大型多序列比对质量的程度。
评估了基于局部序列上下文的两种预测器:(i)单序列二级结构预测,以及(ii)根据周围残基调整空位罚分。结果表明基于上下文的预测器具有可观的信息含量,可用于创建更准确的比对。此外,随着序列数量增加,局部上下文的信息量更大,能够对大型实证基准进行更准确的蛋白质比对。这些发现成为DECIPHER的基础,DECIPHER是一种新的用于序列比对的上下文感知程序,在大型序列集上的表现优于其他程序。
基于局部序列上下文预测二级结构是打破比对中独立性假设的有效手段。由于二级结构比一级序列更保守,可利用它来改善远缘相关蛋白质的比对。此外,随着用于预测的序列增多,二级结构预测准确度提高。这使得能够可扩展地生成大型序列比对,即使在多样的序列集上也能保持高精度。DECIPHER R包和源代码可在DECIPHER.cee.wisc.edu以及Bioconductor仓库免费下载。