Shen Chengze, Liu Baqiao, Williams Kelly P, Warnow Tandy
Computer Science, University of Illinois, Urbana-Champaign, 201 N. Goodwin Ave, Urbana, 61801, IL, USA.
Sandia National Laboratories, 7011 East Ave., Livermore, 94550, CA, USA.
Algorithms Mol Biol. 2023 Dec 7;18(1):21. doi: 10.1186/s13015-023-00247-x.
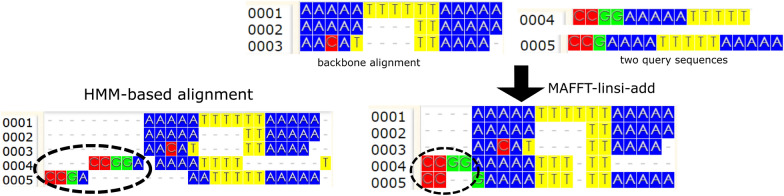
Adding sequences into an existing (possibly user-provided) alignment has multiple applications, including updating a large alignment with new data, adding sequences into a constraint alignment constructed using biological knowledge, or computing alignments in the presence of sequence length heterogeneity. Although this is a natural problem, only a few tools have been developed to use this information with high fidelity.
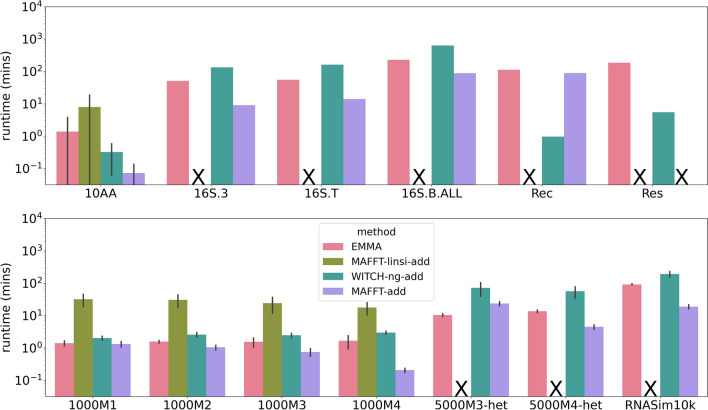
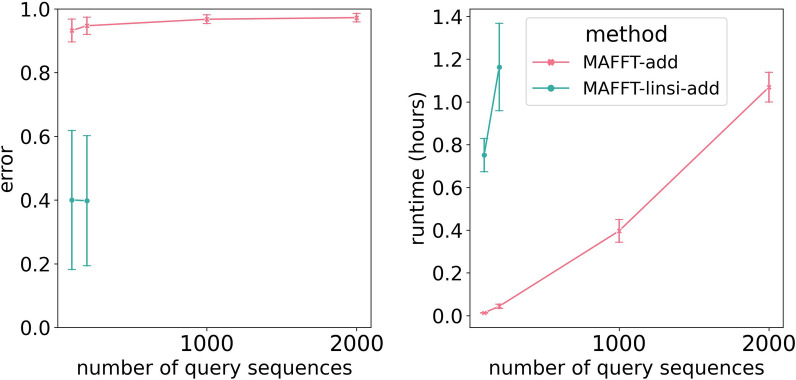
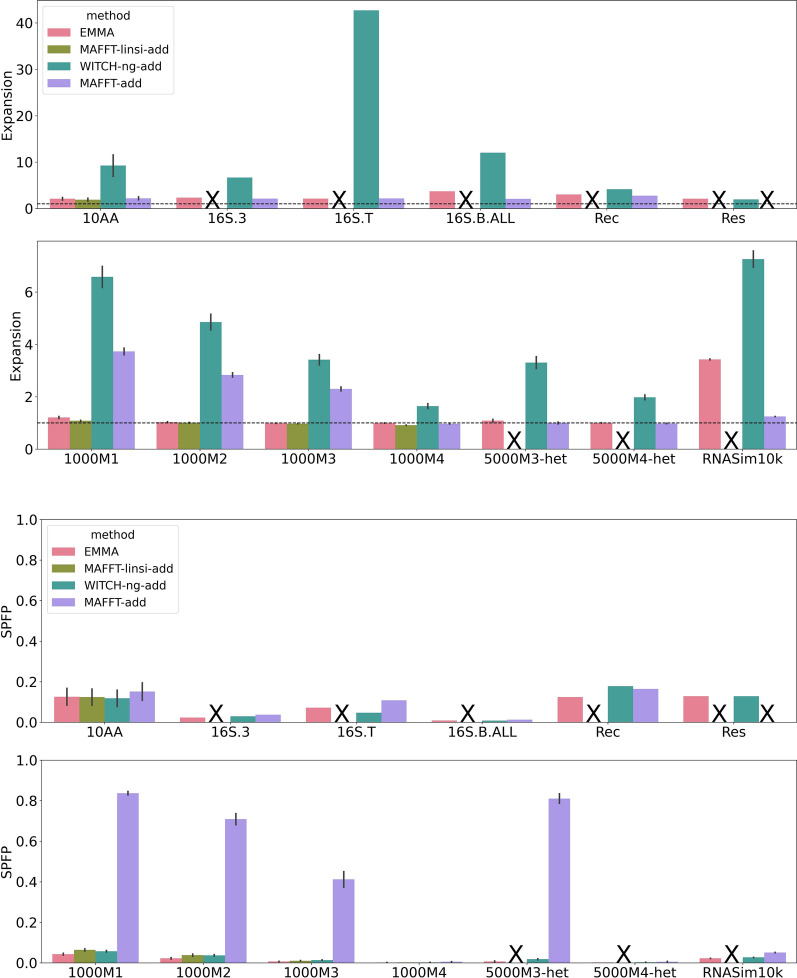
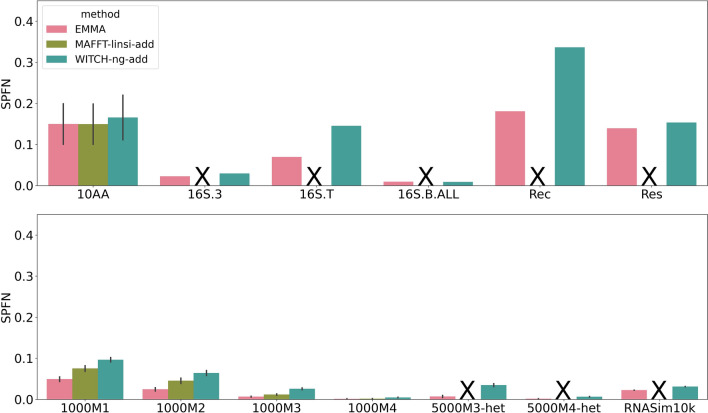
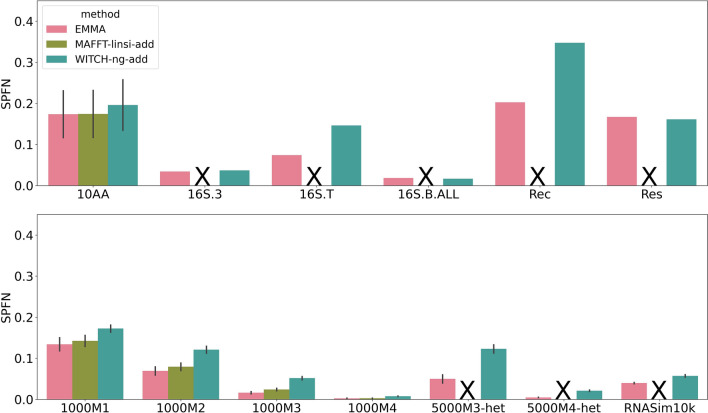
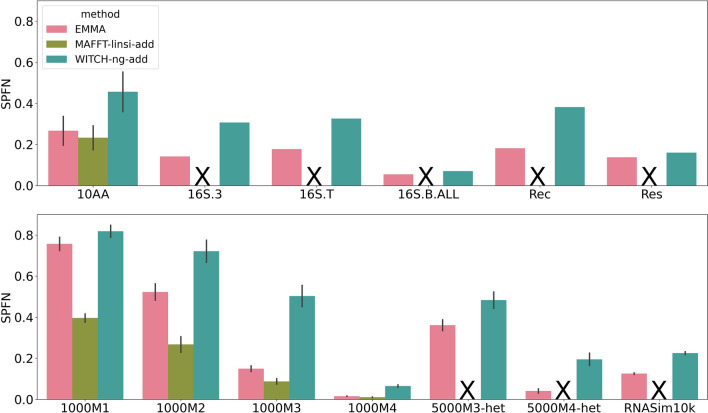
We present EMMA (Extending Multiple alignments using MAFFT--add) for the problem of adding a set of unaligned sequences into a multiple sequence alignment (i.e., a constraint alignment). EMMA builds on MAFFT--add, which is also designed to add sequences into a given constraint alignment. EMMA improves on MAFFT--add methods by using a divide-and-conquer framework to scale its most accurate version, MAFFT-linsi--add, to constraint alignments with many sequences. We show that EMMA has an accuracy advantage over other techniques for adding sequences into alignments under many realistic conditions and can scale to large datasets with high accuracy (hundreds of thousands of sequences). EMMA is available at https://github.com/c5shen/EMMA .
EMMA is a new tool that provides high accuracy and scalability for adding sequences into an existing alignment.
将序列添加到现有的(可能是用户提供的)比对中具有多种应用,包括用新数据更新大型比对、将序列添加到利用生物学知识构建的约束比对中,或在存在序列长度异质性的情况下计算比对。尽管这是一个很自然的问题,但只有少数工具被开发出来以高保真度使用这些信息。
我们提出了EMMA(使用MAFFT扩展多序列比对——添加)来解决将一组未比对序列添加到多序列比对(即约束比对)中的问题。EMMA基于MAFFT——添加构建,MAFFT——添加也是设计用于将序列添加到给定的约束比对中。EMMA通过使用分治框架改进了MAFFT——添加方法,将其最准确的版本MAFFT-linsi——添加扩展到具有许多序列的约束比对。我们表明,在许多实际条件下,EMMA在将序列添加到比对方面比其他技术具有准确性优势,并且可以高精度地扩展到大型数据集(数十万条序列)。EMMA可在https://github.com/c5shen/EMMA获取。
EMMA是一种新工具,为将序列添加到现有比对中提供了高精度和可扩展性。