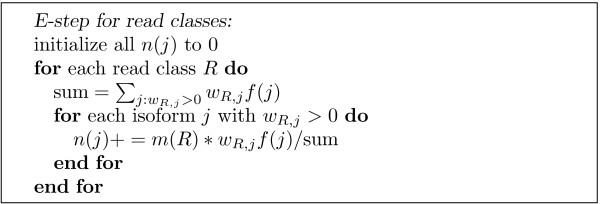Nicolae Marius, Mangul Serghei, Măndoiu Ion I, Zelikovsky Alex
Department of Computer Science & Engineering, University of Connecticut,371 Fairfield Rd,, Unit 2155, Storrs, CT 06269-2155, USA.
Algorithms Mol Biol. 2011 Apr 19;6(1):9. doi: 10.1186/1748-7188-6-9.
Massively parallel whole transcriptome sequencing, commonly referred as RNA-Seq, is quickly becoming the technology of choice for gene expression profiling. However, due to the short read length delivered by current sequencing technologies, estimation of expression levels for alternative splicing gene isoforms remains challenging.
In this paper we present a novel expectation-maximization algorithm for inference of isoform- and gene-specific expression levels from RNA-Seq data. Our algorithm, referred to as IsoEM, is based on disambiguating information provided by the distribution of insert sizes generated during sequencing library preparation, and takes advantage of base quality scores, strand and read pairing information when available. The open source Java implementation of IsoEM is freely available at http://dna.engr.uconn.edu/software/IsoEM/.
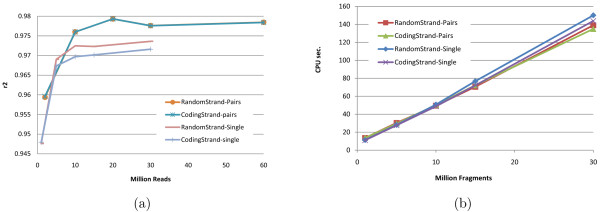
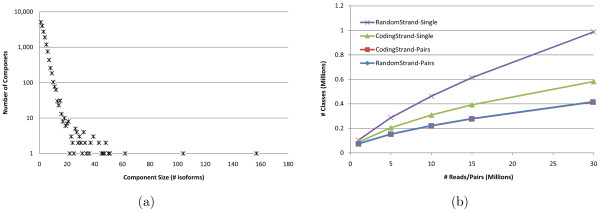
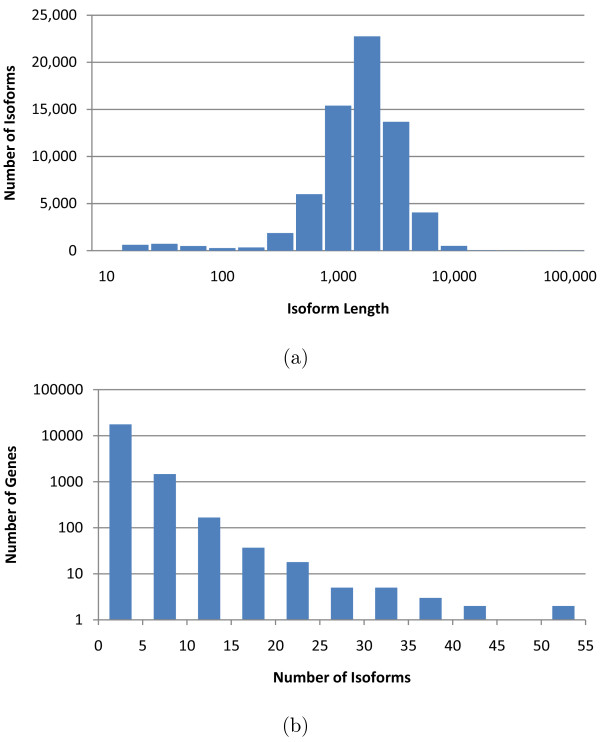
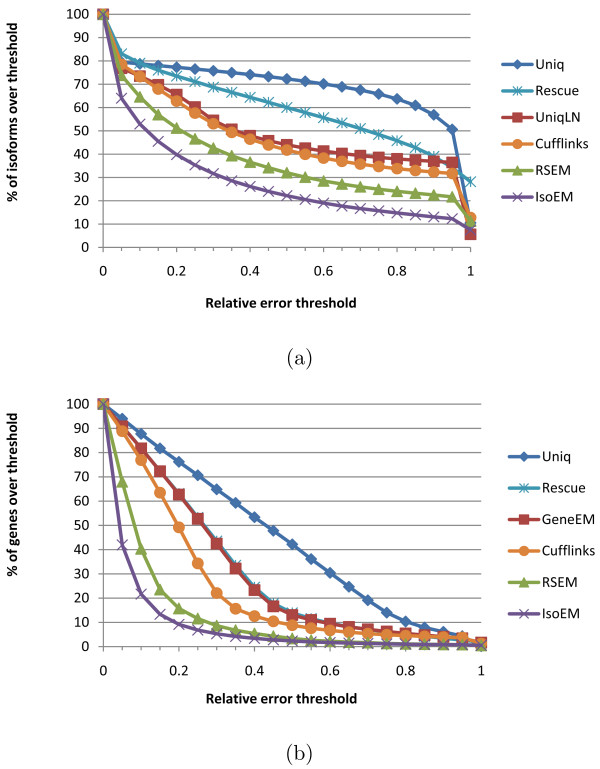
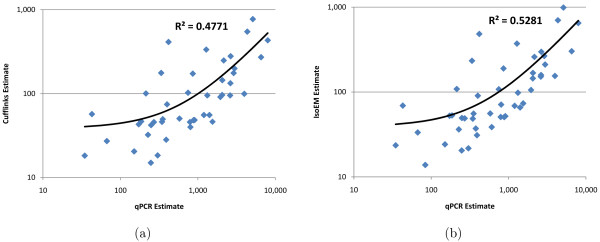
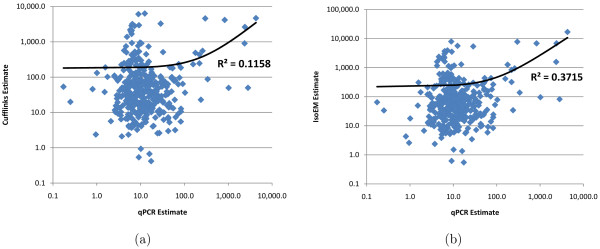
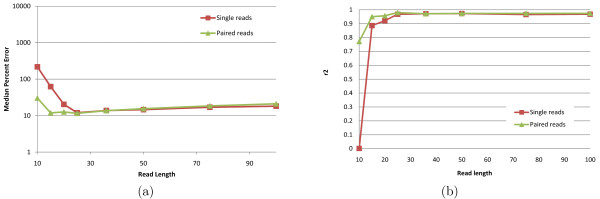
Empirical experiments on both synthetic and real RNA-Seq datasets show that IsoEM has scalable running time and outperforms existing methods of isoform and gene expression level estimation. Simulation experiments confirm previous findings that, for a fixed sequencing cost, using reads longer than 25-36 bases does not necessarily lead to better accuracy for estimating expression levels of annotated isoforms and genes.
大规模平行全转录组测序,通常称为RNA测序(RNA-Seq),正迅速成为基因表达谱分析的首选技术。然而,由于当前测序技术产生的读长较短,对可变剪接基因异构体的表达水平进行估计仍然具有挑战性。
在本文中,我们提出了一种新的期望最大化算法,用于从RNA-Seq数据推断异构体和基因特异性表达水平。我们的算法称为IsoEM,基于对测序文库制备过程中产生的插入片段大小分布所提供的歧义信息进行解析,并在可用时利用碱基质量得分、链和读段配对信息。IsoEM的开源Java实现可从http://dna.engr.uconn.edu/software/IsoEM/免费获取。
对合成和真实RNA-Seq数据集进行的实证实验表明,IsoEM具有可扩展的运行时间,并且在异构体和基因表达水平估计方面优于现有方法。模拟实验证实了先前的发现,即在固定测序成本下,使用长度超过25 - 36个碱基的读段不一定能提高注释异构体和基因表达水平估计的准确性。