Bioinformatics Unit, Institute of Parasitology and Biomedicine "López-Neyra", CSIC (IPBLN-CSIC), 18016 Granada, Spain.
Department of Biochemistry and Molecular Biology, Faculty of Sciences, University of Granada, 18071 Granada, Spain.
Genes (Basel). 2022 Dec 3;13(12):2280. doi: 10.3390/genes13122280.
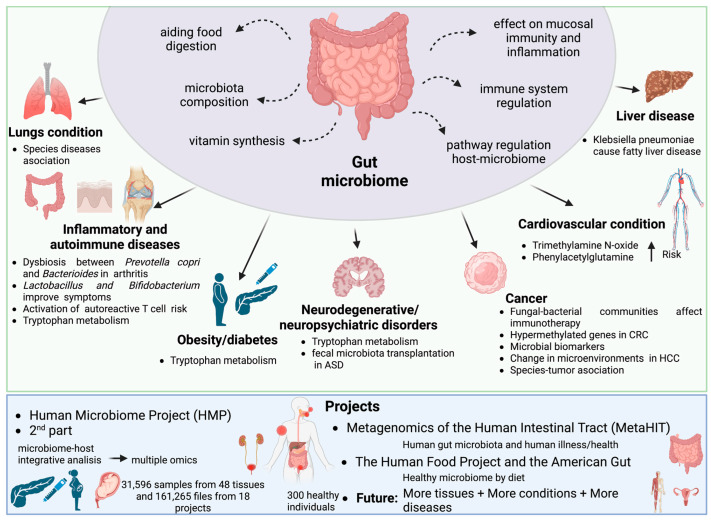
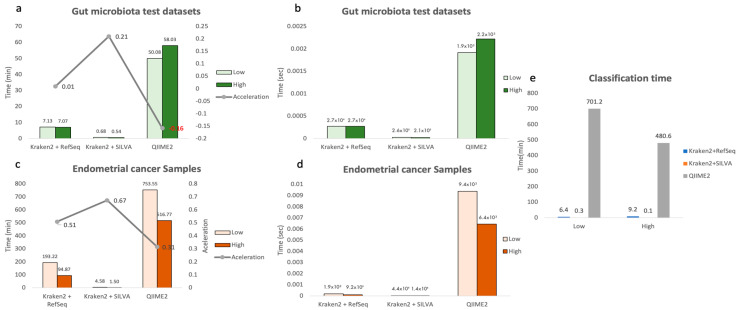
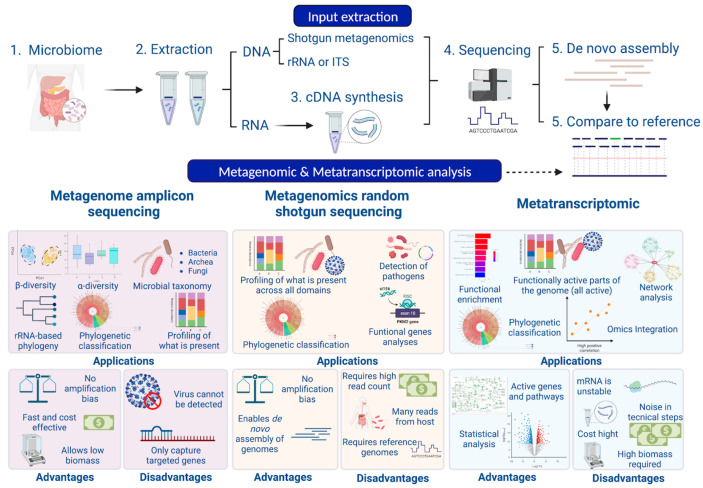
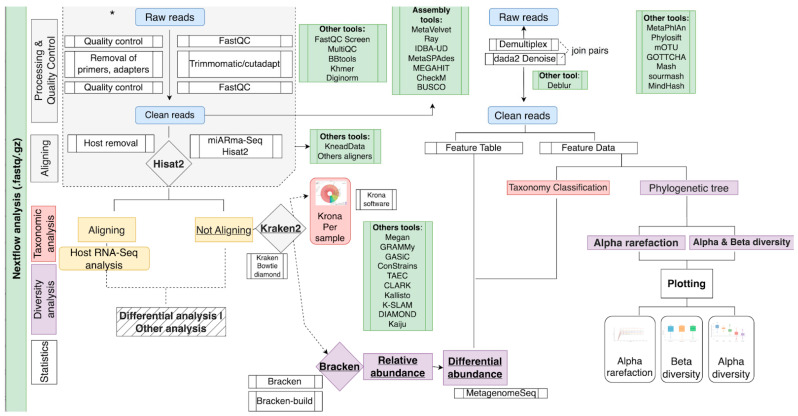
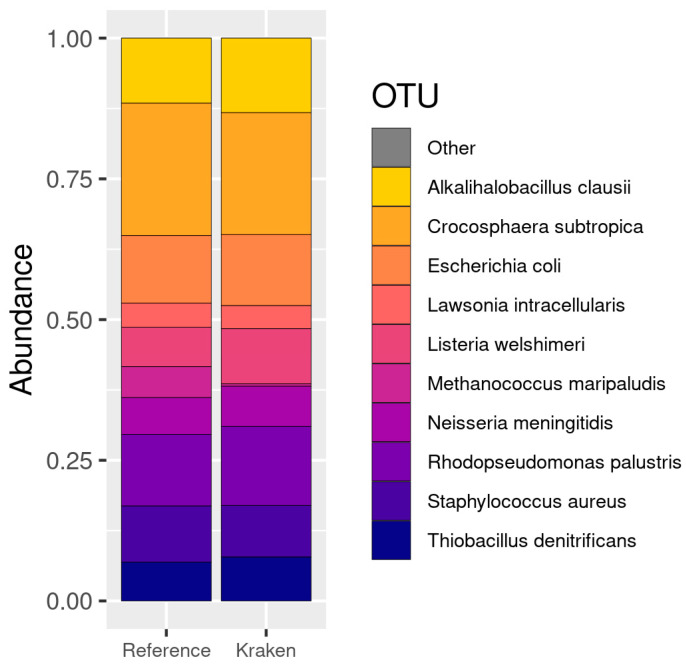
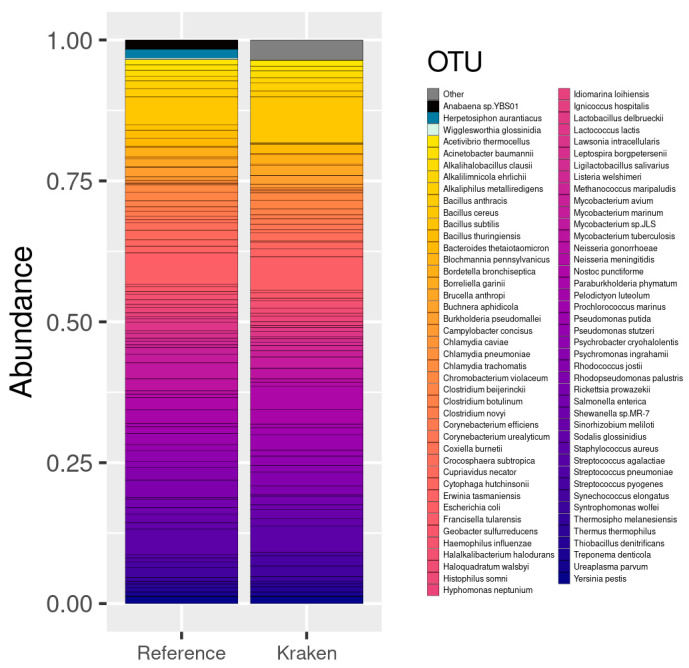
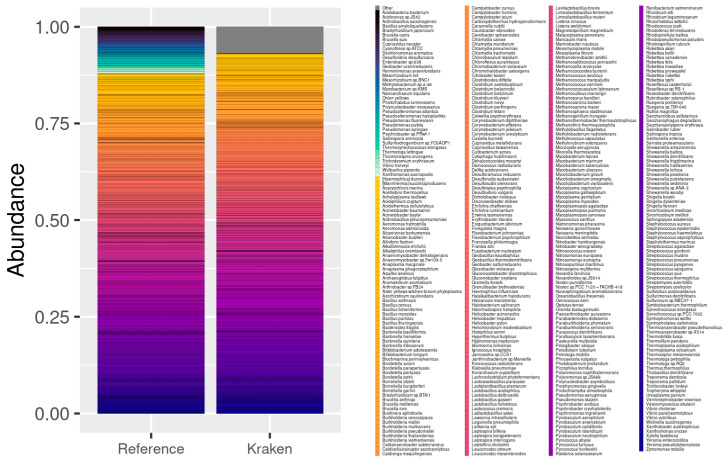
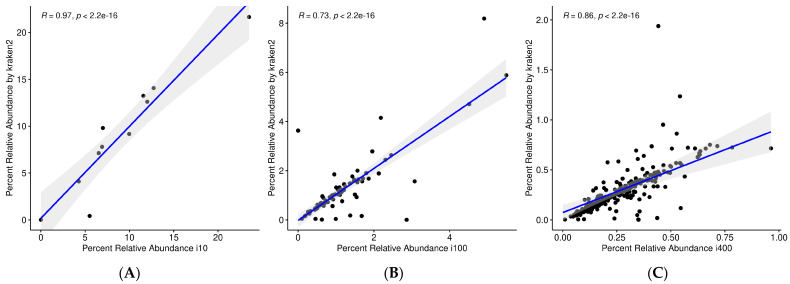
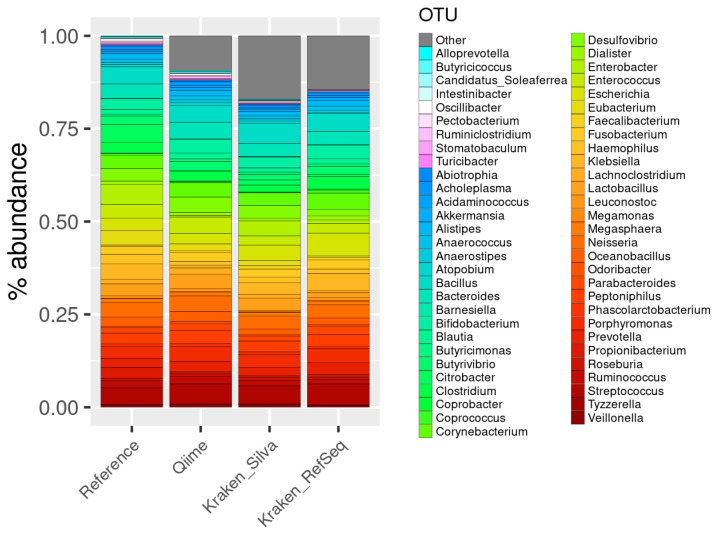
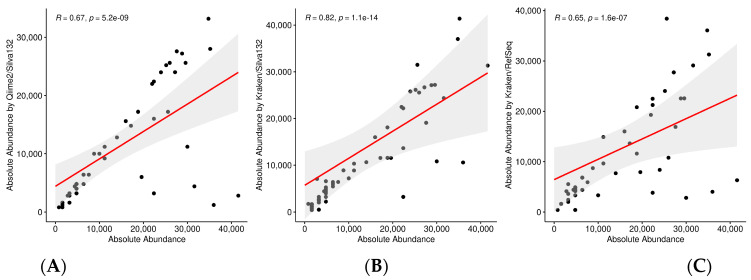
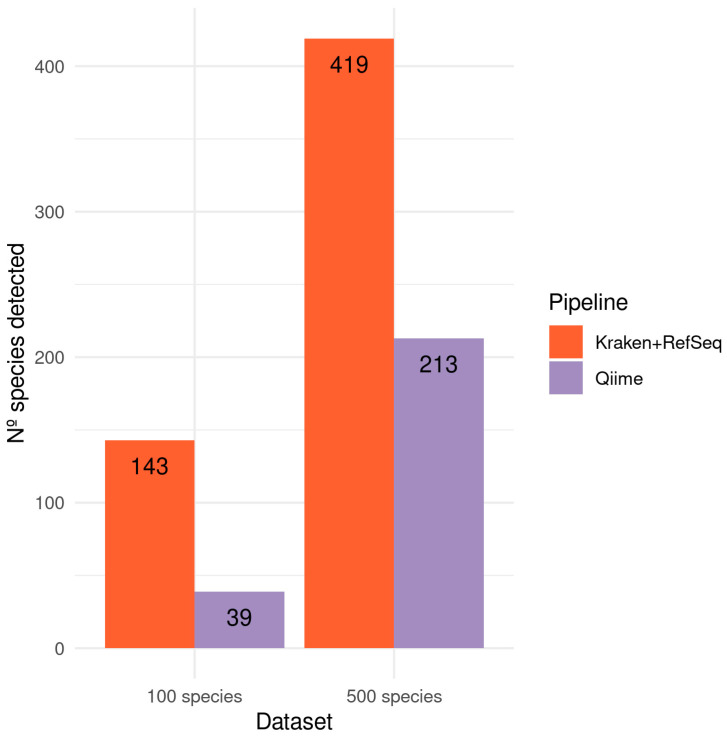
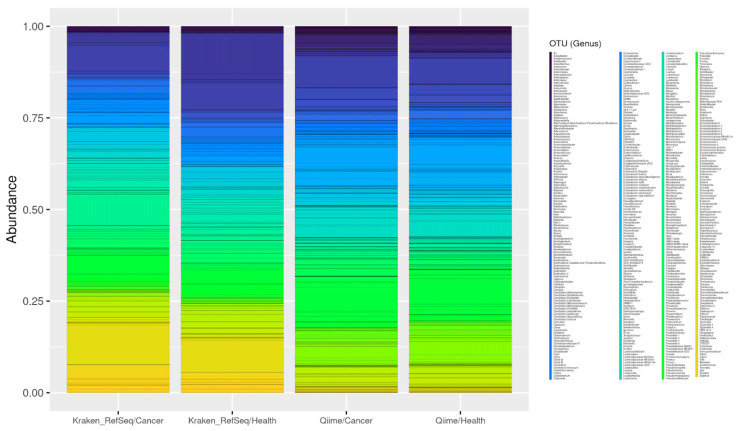
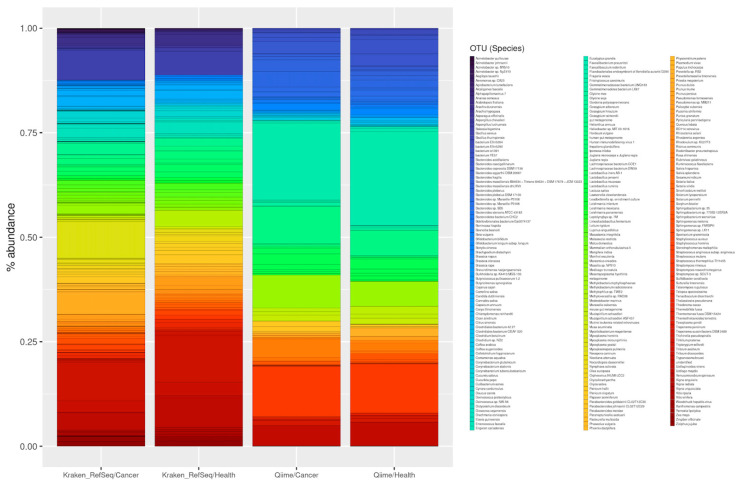
The study of microorganisms is a field of great interest due to their environmental (e.g., soil contamination) and biomedical (e.g., parasitic diseases, autism) importance. The advent of revolutionary next-generation sequencing techniques, and their application to the hypervariable regions of the 16S, 18S or 23S ribosomal subunits, have allowed the research of a large variety of organisms more in-depth, including bacteria, archaea, eukaryotes and fungi. Additionally, together with the development of analysis software, the creation of specific databases (e.g., SILVA or RDP) has boosted the enormous growth of these studies. As the cost of sequencing per sample has continuously decreased, new protocols have also emerged, such as shotgun sequencing, which allows the profiling of all taxonomic domains in a sample. The sequencing of hypervariable regions and shotgun sequencing are technologies that enable the taxonomic classification of microorganisms from the DNA present in microbial communities. However, they are not capable of measuring what is actively expressed. Conversely, we advocate that metatranscriptomics is a "new" technology that makes the identification of the mRNAs of a microbial community possible, quantifying gene expression levels and active biological pathways. Furthermore, it can be also used to characterise symbiotic interactions between the host and its microbiome. In this manuscript, we examine the three technologies above, and discuss the implementation of different software and databases, which greatly impact the obtaining of reliable results. Finally, we have developed two easy-to-use pipelines leveraging Nextflow technology. These aim to provide everything required for an average user to perform a metagenomic analysis of marker genes with QIMME2 and a metatranscriptomic study using Kraken2/Bracken.
由于微生物在环境(例如土壤污染)和生物医学(例如寄生虫病、自闭症)方面的重要性,它们的研究领域引起了极大的兴趣。革命性的下一代测序技术的出现,以及它们在 16S、18S 或 23S 核糖体亚基的高变区的应用,使得对包括细菌、古菌、真核生物和真菌在内的大量生物的研究更加深入。此外,随着分析软件的发展,特定数据库(例如 SILVA 或 RDP)的创建,推动了这些研究的巨大发展。随着每个样本测序成本的持续降低,新的协议也层出不穷,例如 shotgun 测序,它允许对样本中的所有分类域进行分析。高变区测序和 shotgun 测序是从微生物群落中存在的 DNA 对微生物进行分类的技术。然而,它们无法测量活跃表达的内容。相反,我们认为宏转录组学是一种“新技术”,它使得微生物群落的 mRNA 鉴定成为可能,量化基因表达水平和活跃的生物途径。此外,它还可用于描述宿主与其微生物组之间的共生相互作用。在本文中,我们检查了上述三种技术,并讨论了不同软件和数据库的实现,这些都对获得可靠结果产生了重大影响。最后,我们开发了两个利用 Nextflow 技术的易于使用的流程。这些流程旨在为普通用户提供所需的一切,以便使用 QIMME2 对标记基因进行宏基因组分析,并使用 Kraken2/Bracken 进行宏转录组学研究。