Department of Medicine, Washington University, St Louis, Missouri, USA.
JAMA. 2011 Apr 20;305(15):1577-84. doi: 10.1001/jama.2011.497.
Whole-genome sequencing is becoming increasingly available for research purposes, but it has not yet been routinely used for clinical diagnosis.
To determine whether whole-genome sequencing can identify cryptic, actionable mutations in a clinically relevant time frame.
DESIGN, SETTING, AND PATIENT: We were referred a difficult diagnostic case of acute promyelocytic leukemia with no pathogenic X-RARA fusion identified by routine metaphase cytogenetics or interphase fluorescence in situ hybridization (FISH). The case patient was enrolled in an institutional review board-approved protocol, with consent specifically tailored to the implications of whole-genome sequencing. The protocol uses a "movable firewall" that maintains patient anonymity within the entire research team but allows the research team to communicate medically relevant information to the treating physician.
Clinical relevance of whole-genome sequencing and time to communicate validated results to the treating physician.
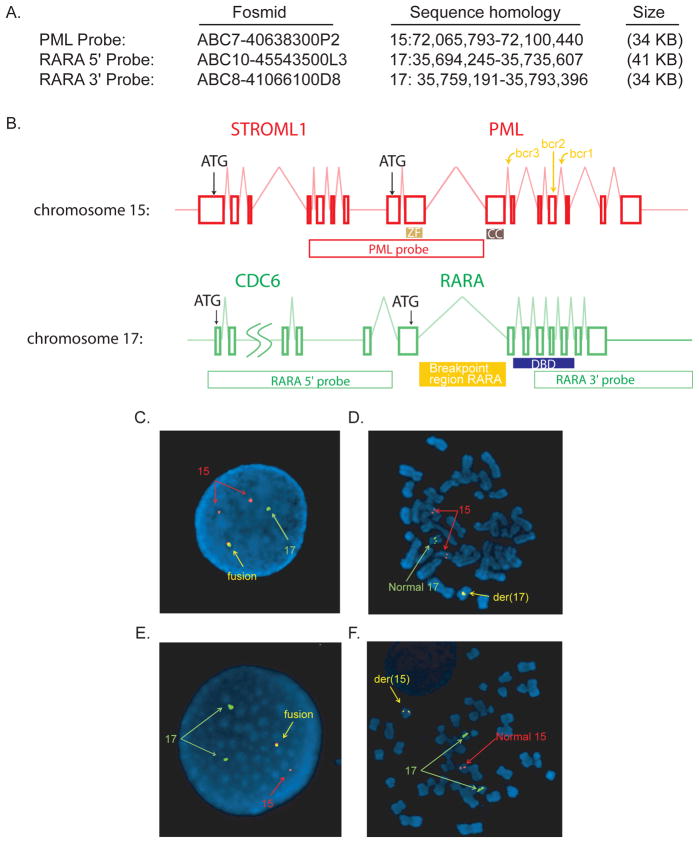
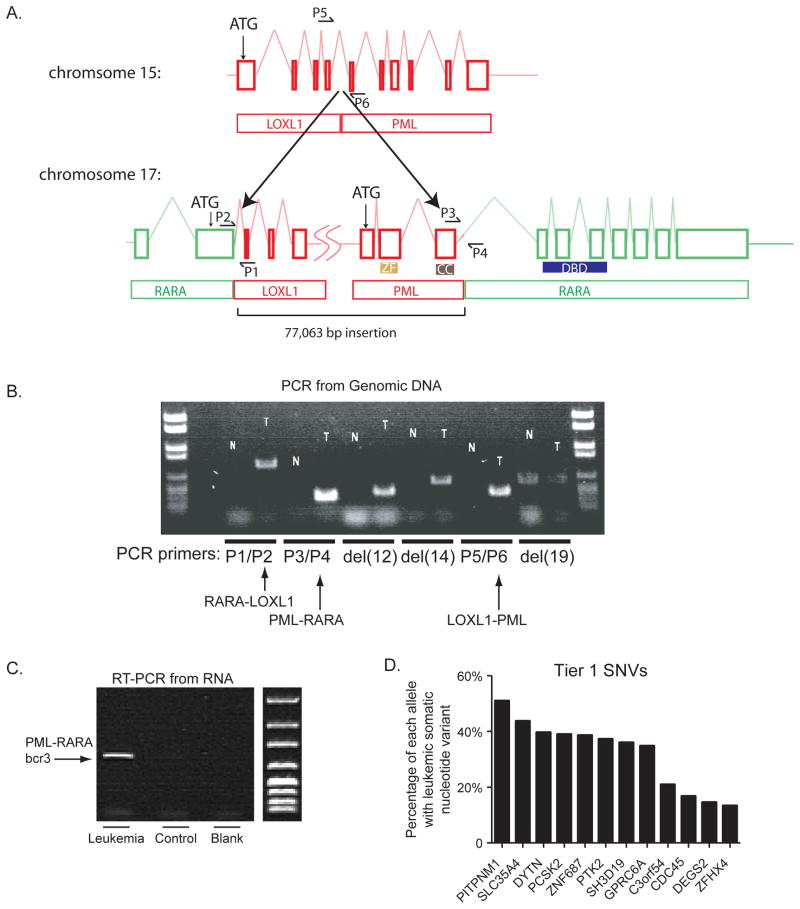
Massively parallel paired-end sequencing allowed identification of a cytogenetically cryptic event: a 77-kilobase segment from chromosome 15 was inserted en bloc into the second intron of the RARA gene on chromosome 17, resulting in a classic bcr3 PML-RARA fusion gene. Reverse transcription polymerase chain reaction sequencing subsequently validated the expression of the fusion transcript. Novel FISH probes identified 2 additional cases of t(15;17)-negative acute promyelocytic leukemia that had cytogenetically invisible insertions. Whole-genome sequencing and validation were completed in 7 weeks and changed the treatment plan for the patient.
Whole-genome sequencing can identify cytogenetically invisible oncogenes in a clinically relevant time frame.
全基因组测序在研究目的上越来越普及,但尚未常规用于临床诊断。
确定全基因组测序是否能在临床相关的时间范围内识别隐匿性、可操作的突变。
设计、设置和患者:我们被转介了一个具有挑战性的急性早幼粒细胞白血病病例,通过常规中期细胞遗传学或间期荧光原位杂交(FISH)均未发现致病性 X-RARA 融合。该病例患者被纳入机构审查委员会批准的方案,同意书专门针对全基因组测序的影响进行了定制。该方案使用了“可移动防火墙”,在整个研究团队中维护患者的匿名性,但允许研究团队将与医学相关的信息传达给主治医生。
全基因组测序的临床相关性和向主治医生传达验证结果的时间。
大规模平行配对末端测序能够识别出一种细胞遗传学上隐匿的事件:来自 15 号染色体的 77 千碱基片段整体插入 17 号染色体上的 RARA 基因的第二个内含子,导致经典的 bcr3 PML-RARA 融合基因。随后,逆转录聚合酶链反应测序验证了融合转录本的表达。新型 FISH 探针鉴定出另外 2 例细胞遗传学上不可见的 15;17 易位阴性急性早幼粒细胞白血病,这些白血病具有细胞遗传学上不可见的插入。全基因组测序和验证在 7 周内完成,并改变了患者的治疗计划。
全基因组测序可以在临床相关的时间范围内识别细胞遗传学上不可见的致癌基因。