Frimayanti Neni, Chee Chin Fei, Zain Sharifuddin M, Rahman Noorsaadah Abd
Department of Chemistry, Faculty of Science, University of Malaya, 50603 Lembah Pantai, Kuala Lumpur, Malaysia; E-Mails:
Int J Mol Sci. 2011 Feb 9;12(2):1089-100. doi: 10.3390/ijms12021089.
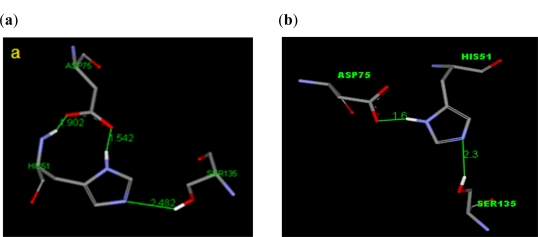
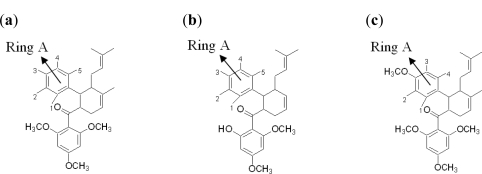
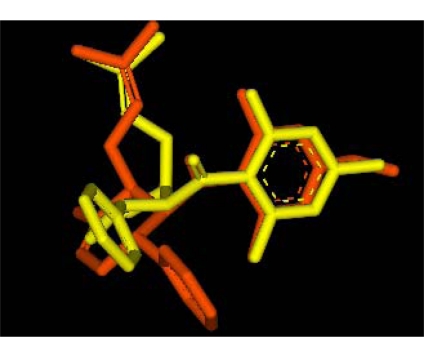
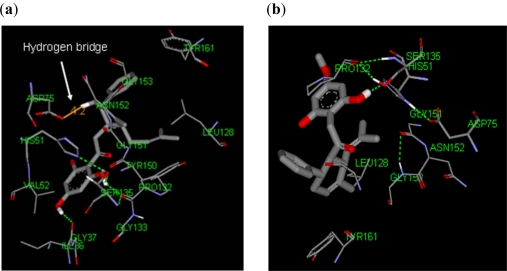
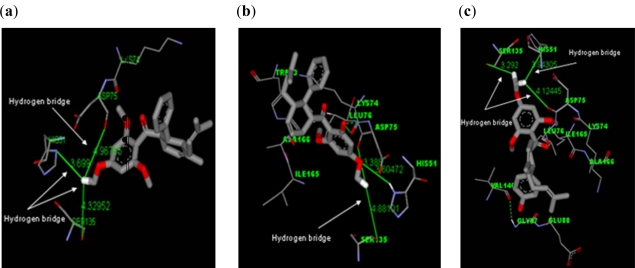
Dengue is a serious disease which has become a global health burden in the last decade. Currently, there are no approved vaccines or antiviral therapies to combat the disease. The increasing spread and severity of the dengue virus infection emphasizes the importance of drug discovery strategies that could efficiently and cost-effectively identify antiviral drug leads for development into potent drugs. To this effect, several computational approaches were applied in this work. Initially molecular docking studies of reference ligands to the DEN2 NS2B/NS3 serine protease were carried out. These reference ligands consist of reported competitive inhibitors extracted from Boesenbergia rotunda (i.e., 4-hydroxypanduratin A and panduratin A) and three other synthesized panduratin A derivative compounds (i.e., 246DA, 2446DA and 20H46DA). The design of new lead inhibitors was carried out in two stages. In the first stage, the enzyme complexed to the reference ligands was minimized and their complexation energies (i.e., sum of interaction energy and binding energy) were computed. New compounds as potential dengue inhibitors were then designed by putting various substituents successively on the benzyl ring A of the reference molecule. These substituted benzyl compounds were then computed for their enzyme-ligand complexation energies. New enzyme-ligand complexes, exhibiting the lowest complexation energies and closest to the computed energy for the reference compounds, were then chosen for the next stage manipulation and design, which involved substituting positions 4 and 5 of the benzyl ring A (positions 3 and 4 for 2446DA) with various substituents.
登革热是一种严重的疾病,在过去十年中已成为全球健康负担。目前,尚无经批准的疫苗或抗病毒疗法来对抗这种疾病。登革热病毒感染的传播范围和严重程度不断增加,凸显了药物发现策略的重要性,这些策略能够高效且经济地识别抗病毒药物先导物,以开发成强效药物。为此,本研究采用了几种计算方法。首先对参考配体与DEN2 NS2B/NS3丝氨酸蛋白酶进行了分子对接研究。这些参考配体包括从圆叶姜中提取的已报道的竞争性抑制剂(即4-羟基潘多拉丁A和潘多拉丁A)以及其他三种合成的潘多拉丁A衍生物化合物(即246DA、2446DA和20H46DA)。新型先导抑制剂的设计分两个阶段进行。在第一阶段,将与参考配体复合的酶进行最小化处理,并计算它们的复合能(即相互作用能和结合能之和)。然后通过在参考分子的苄基环A上依次引入各种取代基来设计作为潜在登革热抑制剂的新化合物。接着计算这些取代苄基化合物的酶-配体复合能。然后选择具有最低复合能且最接近参考化合物计算能量的新型酶-配体复合物进行下一阶段的操作和设计,这涉及用各种取代基取代苄基环A的4位和5位(2446DA为3位和4位)。