Center for Metabolic Bone Disease and Molecular Research, Shriners Hospital for Children, St. Louis, MO 63131, USA.
J Bone Miner Res. 2011 May;26(5):920-33. doi: 10.1002/jbmr.283.
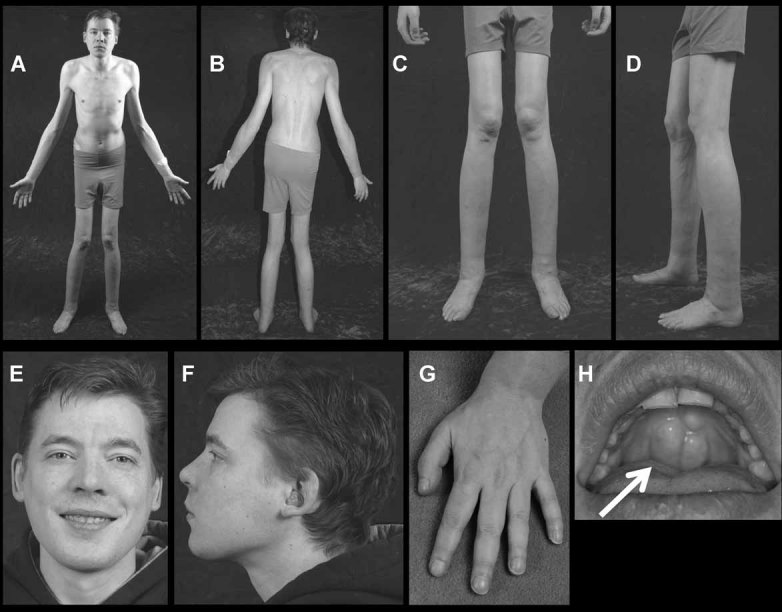
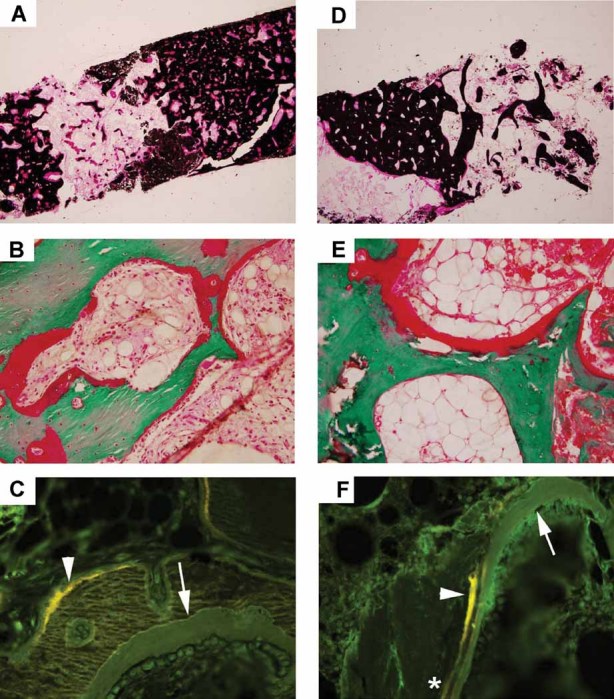
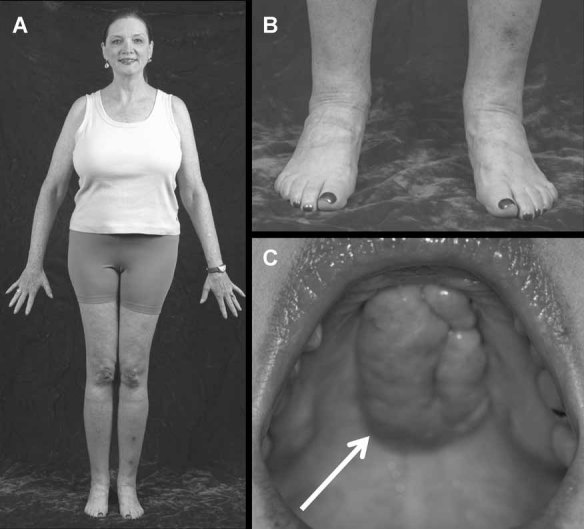
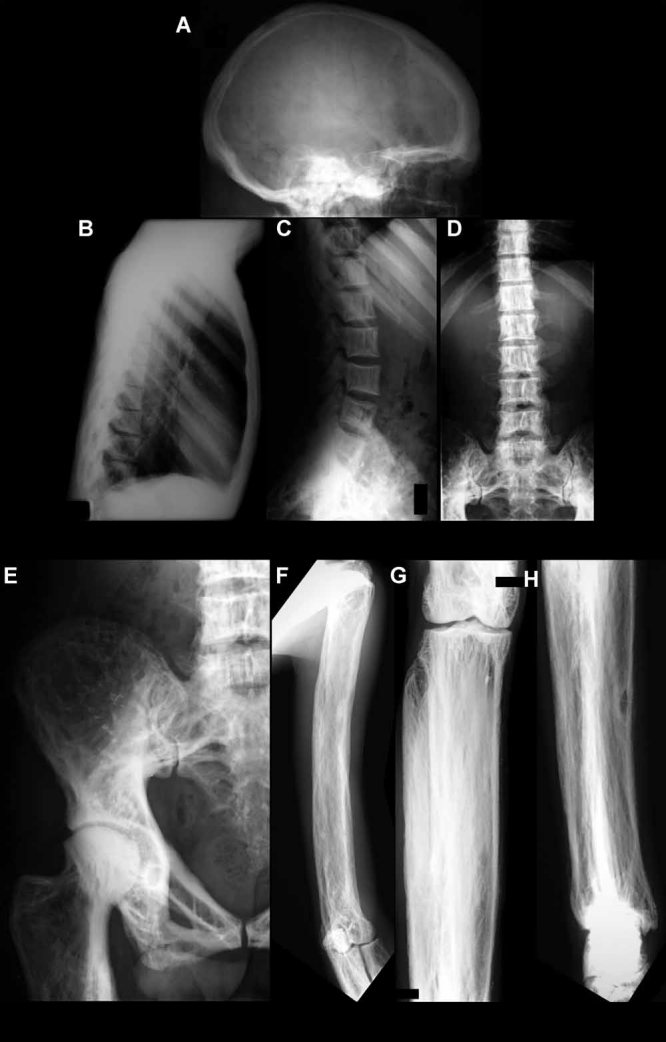
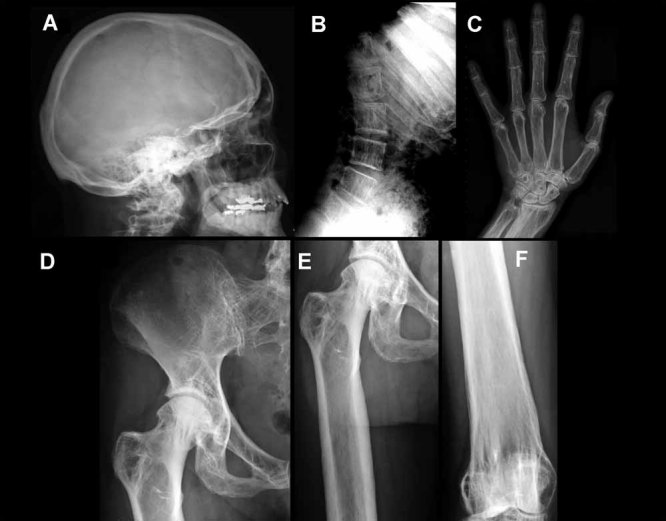
We report a 32-year-old man and his 59-year-old mother with a unique and extensive variant of Camurati-Engelmann disease (CED) featuring histopathological changes of osteomalacia and alterations within TGFβ1 and TNFSF11 encoding TGFβ1 and RANKL, respectively. He suffered leg pain and weakness since childhood and reportedly grew until his late 20s, reaching 7 feet in height. He had deafness, perforated nasal septum, torus palatinus, disproportionately long limbs with knock-knees, low muscle mass, and pseudoclubbing. Radiographs revealed generalized skeletal abnormalities, including wide bones and cortical and trabecular bone thickening in keeping with CED, except that long bone ends were also affected. Lumbar spine and hip BMD Z-scores were + 7.7 and + 4.4, respectively. Biochemical markers of bone turnover were elevated. Hypocalciuria accompanied low serum 25-hydroxyvitamin D (25[OH]D) levels. Pituitary hypogonadism and low serum insulin-like growth factor (IGF)-1 were present. Karyotype was normal. Despite vitamin D repletion, iliac crest histology revealed severe osteomalacia. Exon 1 of TNFRSF11A (RANK), exons 2, 3, and 4 of LRP5, and all coding exons and adjacent mRNA splice junctions of TNFRSF11B (OPG), SQSTM1 (sequestosome 1), and TNSALP (tissue nonspecific alkaline phosphatase) were intact. His asymptomatic and less dysmorphic 5'11″ mother, also with low serum 25(OH)D, had milder clinical, radiological, biochemical, and histopathological findings. Both individuals were heterozygous for a novel 12-bp duplication (c.27_38dup, p.L10_L13dup) in exon 1 of TGFβ1, predicting four additional leucine residues in the latency-associated-peptide segment of TGFβ1, consistent with CED. The son was also homozygous for a single base transversion in TNFSF11, predicting a nonconservative amino acid change (c.107C > G, p.Pro36Arg) in the intracellular domain of RANKL that was heterozygous in his nonconsanguineous parents. This TNFSF11 variant was not found in the SNP Database, nor in published TNFSF11 association studies, but it occurred in four of the 134 TNFSF11 alleles (3.0%) we tested randomly among individuals without CED. Perhaps the unique phenotype of this CED family is conditioned by altered RANKL activity.
我们报告了一例 32 岁男性及其 59 岁母亲,他们患有卡穆拉蒂-恩格尔曼病(CED)的独特且广泛变异型,其特征为骨软化症的组织病理学变化以及 TGFβ1 和 TNFSF11 编码 TGFβ1 和 RANKL 的改变。他从小就患有腿部疼痛和无力,据报道他的生长持续到 20 多岁,身高达到 7 英尺。他患有耳聋、鼻中隔穿孔、腭弓、不成比例的长肢伴膝内翻、肌肉量低和假性杵状指。X 线片显示全身性骨骼异常,包括宽骨和皮质及骨小梁增厚,符合 CED,但长骨末端也受到影响。腰椎和髋关节 BMD Z 评分分别为+7.7 和+4.4。骨转换生化标志物升高。低钙尿症伴有低血清 25-羟维生素 D(25[OH]D)水平。存在垂体性腺功能减退和低血清胰岛素样生长因子(IGF)-1。核型正常。尽管补充了维生素 D,但髂嵴组织学显示严重的骨软化症。TNFRSF11A(RANK)的外显子 1、LRP5 的外显子 2、3 和 4 以及 TNFRSF11B(OPG)、SQSTM1(自噬体 1)和 TNSALP(组织非特异性碱性磷酸酶)的所有编码外显子和相邻 mRNA 剪接接头均完整。他无症状且畸形较轻的 5'11″母亲,血清 25(OH)D 也较低,其临床表现、影像学、生化和组织病理学发现较轻微。这两个人都是 TGFβ1 外显子 1 中 12 个碱基对重复(c.27_38dup,p.L10_L13dup)的杂合子,预测 TGFβ1 潜伏期相关肽段中会有四个额外的亮氨酸残基,与 CED 一致。儿子也是 TNFSF11 中单个碱基颠换的纯合子,预测 RANKL 细胞内结构域中存在非保守氨基酸变化(c.107C > G,p.Pro36Arg),其父母为杂合子。该 TNFSF11 变体未在 SNP 数据库中找到,也未在已发表的 TNFSF11 关联研究中找到,但在我们随机测试的 134 个无 CED 的个体中,该 TNFSF11 等位基因的四个中发现了(3.0%)。也许这个 CED 家族的独特表型是由改变的 RANKL 活性决定的。