Department of Medical Informatics and Clinical Epidemiology, Oregon Health & Science University, 3181 SW Sam Jackson Park Road, Portland, Oregon 97239, USA.
J Proteome Res. 2011 Jul 1;10(7):2905-12. doi: 10.1021/pr200133p. Epub 2011 May 9.
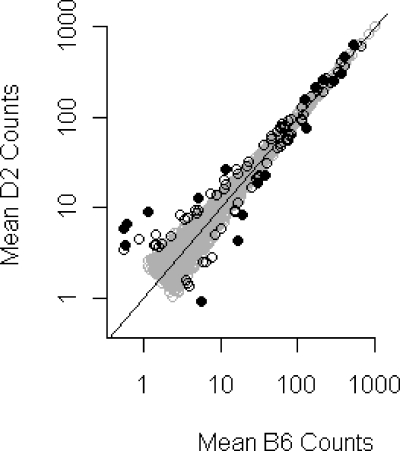
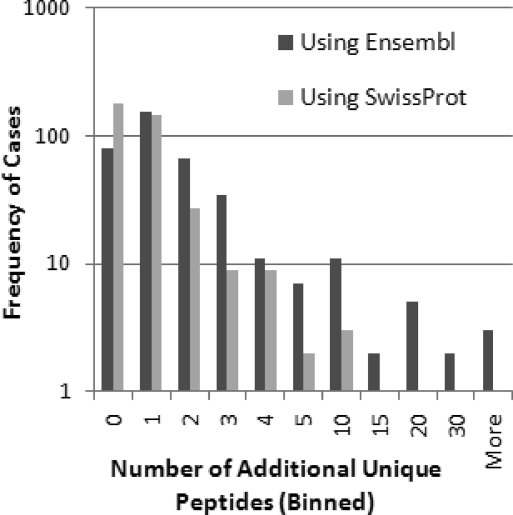
Decades of genetics research comparing mouse strains has identified many regions of the genome associated with quantitative traits. Microarrays have been used to identify which genes in those regions are differentially expressed and are therefore potentially causal; however, genetic variants that affect probe hybridization lead to many false conclusions. Here we used spectral counting to compare brain striata between two mouse strains. Using strain-specific protein databases, we concluded that proteomics was more robust to sequence differences than microarrays; however, some proteins were still significantly affected. To generate strain-specific databases, we used a complete database that contained all of the putative genetic isoforms for each protein. While the increased proteome coverage in the databases led to a 6.8% gain in peptide assignments compared to a nonredundant database, it also necessitated the development of a strategy for grouping similar proteins due to a large number of shared peptides. Of the 4563 identified proteins (2.1% FDR), there were 1807 quantifiable proteins/groups that exceeded minimum count cutoffs. With four pooled biological replicates per strain, we used quantile normalization, ComBat (a package that adjusts for batch effects), and edgeR (a package for differential expression analysis of count data) to identify 101 differentially expressed proteins/groups, 84 of which had a coding region within one of the genomic regions of interest identified by the Portland Alcohol Research Center.
几十年来,对老鼠品系进行的遗传学研究已经确定了与数量性状相关的基因组的许多区域。微阵列已被用于识别那些区域中差异表达的基因,因此这些基因可能是潜在的原因;然而,影响探针杂交的遗传变异会导致许多错误的结论。在这里,我们使用光谱计数法比较了两种小鼠品系的大脑纹状体。使用特定于品系的蛋白质数据库,我们得出结论,蛋白质组学比微阵列更能抵抗序列差异;然而,一些蛋白质仍然受到显著影响。为了生成特定于品系的数据库,我们使用了一个包含每个蛋白质所有假定遗传同工型的完整数据库。虽然数据库中增加的蛋白质组覆盖范围导致肽分配增加了 6.8%,与非冗余数据库相比,但由于大量共享肽,也需要开发一种对相似蛋白质进行分组的策略。在鉴定出的 4563 种蛋白质(2.1% FDR)中,有 1807 种可量化蛋白质/组超过最小计数阈值。对于每一种品系的四个混合生物重复,我们使用分位数归一化、ComBat(一种用于调整批次效应的软件包)和 edgeR(一种用于对计数数据进行差异表达分析的软件包)来识别 101 个差异表达的蛋白质/组,其中 84 个蛋白质的编码区位于波特兰酒精研究中心确定的一个感兴趣的基因组区域内。