Matsuo Teppei, Nishizuka Satoshi S, Ishida Kazushige, Iwaya Takeshi, Ikeda Miyuki, Wakabayashi Go
Molecular Therapeutics Laboratory, Department of Surgery, Iwate Medical University School of Medicine, 19-1 Uchimaru, Morioka 020-8505, Iwate Japan.
BMC Res Notes. 2011 May 10;4:140. doi: 10.1186/1756-0500-4-140.
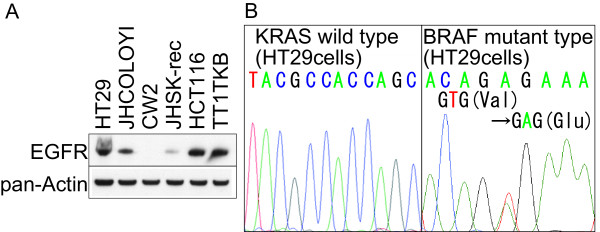
The binding of EGFR and its ligands leads to autophosphorylation of receptor tyrosine kinase as well as subsequent activation of signal transduction pathways that are involved in regulating cellular proliferation, differentiation, and survival. An EGFR inhibitor, cetuximab binds to EGFR and consequently blocks a variety of cellular processes. KRAS/BRAF mutations are known to be associated with a low response rate to cetuximab. In the present study, to clarify the anti-tumor mechanisms of cetuximab, we evaluated the KRAS/BRAF status, phosphorylation level of the EGFR pathway, and the tumor suppression effect in vivo, using a human colon cancer cell line HT29, which exhibited the highest EGFR expression in response to the cetuximab therapy among the 6 colorectal cancer cell lines tested.
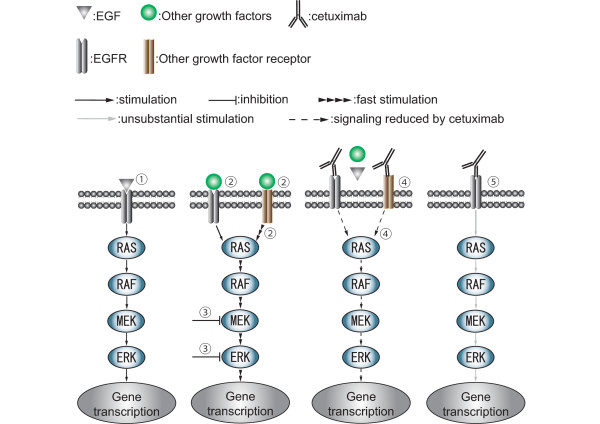
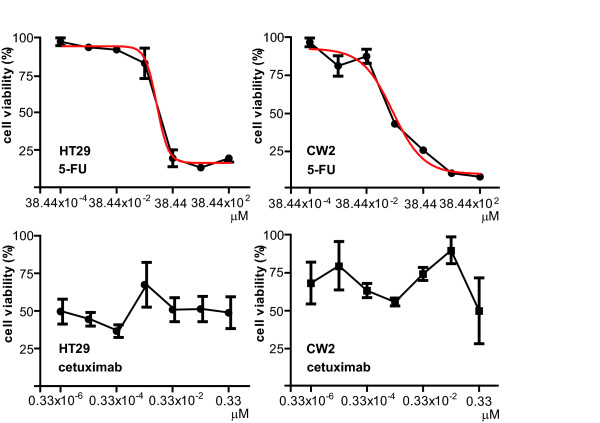
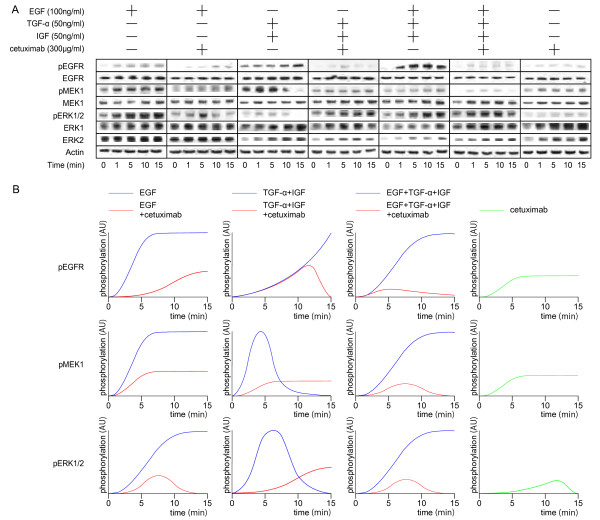
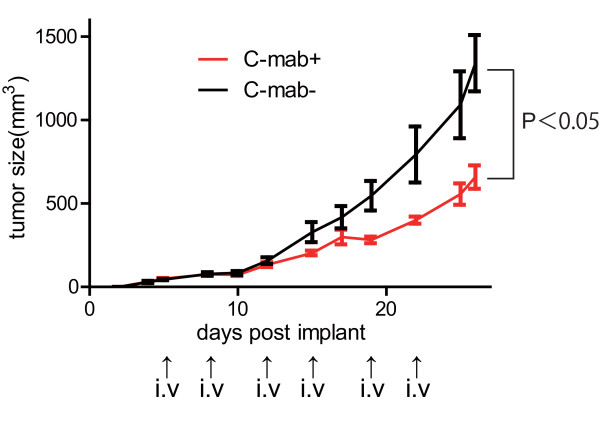
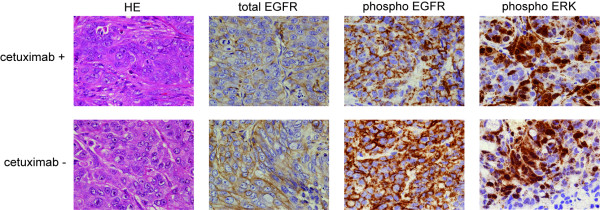
The conventional growth suppression assay did not work efficiently with cetuximab. EGF, TGF-α, and IGF activated the EGFR/MAPK cell signaling pathway by initiating the phosphorylation of EGFR. Cetuximab partially inhibited the EGFR/MAPK pathway induced by EGF, TGF-α, and IGF. However, cetuximab exposure induced the EGFR, MEK, and ERK1/2 phosphorylation by itself. Mouse xenograft tumor growth was significantly inhibited by cetuximab and both cetuximab-treated and -untreated xenograft specimens exhibited phosphorylations of the EGFR pathway proteins.
We have confirmed that cetuximab inhibited the EGFR/MAPK pathway and reduced tumor growth in the xenografts while the remaining tumor showed EGFR pathway activation. These results suggest that: ( i ) The effect of cetuximab in growth signaling is not sufficient to induce complete growth suppression in vitro; ( ii ) time-course monitoring may be necessary to evaluate the effect of cetuximab because EGFR signaling is transmitted in a minute order; and ( iii ) cetuximab treatment may have cells acquired resistant selectively survived in the heterogeneous cancer population.
表皮生长因子受体(EGFR)与其配体的结合会导致受体酪氨酸激酶的自磷酸化,以及随后信号转导通路的激活,这些信号转导通路参与调节细胞增殖、分化和存活。一种EGFR抑制剂西妥昔单抗可与EGFR结合,从而阻断多种细胞过程。已知KRAS/BRAF突变与对西妥昔单抗的低反应率相关。在本研究中,为阐明西妥昔单抗的抗肿瘤机制,我们使用人结肠癌细胞系HT29评估了KRAS/BRAF状态、EGFR通路的磷酸化水平以及体内肿瘤抑制效果。在测试的6种结肠癌细胞系中,HT29对西妥昔单抗治疗的EGFR表达最高。
传统的生长抑制试验对西妥昔单抗效果不佳。表皮生长因子(EGF)、转化生长因子-α(TGF-α)和胰岛素样生长因子(IGF)通过启动EGFR的磷酸化来激活EGFR/丝裂原活化蛋白激酶(MAPK)细胞信号通路。西妥昔单抗部分抑制了由EGF、TGF-α和IGF诱导的EGFR/MAPK通路。然而,西妥昔单抗自身暴露会诱导EGFR、丝裂原活化蛋白激酶激酶(MEK)和细胞外信号调节激酶1/2(ERK1/2)的磷酸化。西妥昔单抗显著抑制了小鼠异种移植瘤的生长,且接受西妥昔单抗治疗和未治疗的异种移植标本均显示出EGFR通路蛋白的磷酸化。
我们已证实西妥昔单抗抑制了EGFR/MAPK通路并减少了异种移植瘤的生长,而剩余肿瘤显示出EGFR通路激活。这些结果表明:(i)西妥昔单抗在生长信号传导方面的作用不足以在体外诱导完全的生长抑制;(ii)由于EGFR信号是按微小顺序传递的,可能需要进行时间进程监测来评估西妥昔单抗的效果;(iii)西妥昔单抗治疗可能使在异质性癌群体中获得抗性的细胞选择性存活。