Genome Sciences Centre, British Columbia Cancer Agency, Vancouver, British Columbia, Canada.
PLoS One. 2011 May 11;6(5):e19816. doi: 10.1371/journal.pone.0019816.
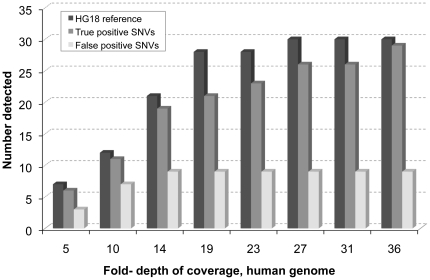
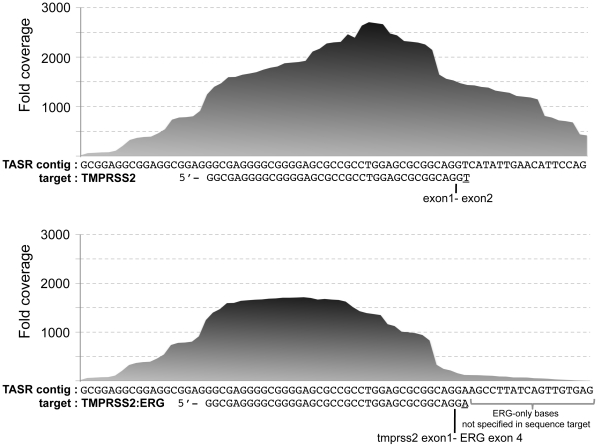
As next-generation sequence (NGS) production continues to increase, analysis is becoming a significant bottleneck. However, in situations where information is required only for specific sequence variants, it is not necessary to assemble or align whole genome data sets in their entirety. Rather, NGS data sets can be mined for the presence of sequence variants of interest by localized assembly, which is a faster, easier, and more accurate approach. We present TASR, a streamlined assembler that interrogates very large NGS data sets for the presence of specific variants by only considering reads within the sequence space of input target sequences provided by the user. The NGS data set is searched for reads with an exact match to all possible short words within the target sequence, and these reads are then assembled stringently to generate a consensus of the target and flanking sequence. Typically, variants of a particular locus are provided as different target sequences, and the presence of the variant in the data set being interrogated is revealed by a successful assembly outcome. However, TASR can also be used to find unknown sequences that flank a given target. We demonstrate that TASR has utility in finding or confirming genomic mutations, polymorphisms, fusions and integration events. Targeted assembly is a powerful method for interrogating large data sets for the presence of sequence variants of interest. TASR is a fast, flexible and easy to use tool for targeted assembly.
随着下一代测序(NGS)的产量不断增加,分析已成为一个重大瓶颈。然而,在仅需要特定序列变异信息的情况下,没有必要对整个基因组数据集进行组装或对齐。相反,可以通过局部组装来挖掘 NGS 数据集以查找感兴趣的序列变异,这是一种更快、更容易、更准确的方法。我们提出了 TASR,这是一种精简的组装程序,通过仅考虑用户提供的输入目标序列的序列空间内的读取,来检测非常大的 NGS 数据集是否存在特定变体。在 NGS 数据集中搜索与目标序列内所有可能的短字完全匹配的读取,然后严格组装这些读取,以生成目标和侧翼序列的共识。通常,特定位置的变体作为不同的目标序列提供,并且通过成功的组装结果揭示了正在检测的数据集中变体的存在。然而,TASR 也可用于查找给定目标侧翼的未知序列。我们证明 TASR 在发现或确认基因组突变、多态性、融合和整合事件方面具有实用性。靶向组装是一种用于检测大型数据集是否存在感兴趣的序列变异的强大方法。TASR 是一种快速、灵活且易于使用的靶向组装工具。