Department of Medicine, Diabetes Center, University of California San Francisco, San Francisco, California, United States of America.
PLoS One. 2011 May 11;6(5):e19878. doi: 10.1371/journal.pone.0019878.
The pathogenesis of insulin resistance in the absence of obesity is unknown. In obesity, multiple stress kinases have been identified that impair the insulin signaling pathway via serine phosphorylation of key second messenger proteins. These stress kinases are activated through various mechanisms related to lipid oversupply locally in insulin target tissues and in various adipose depots.
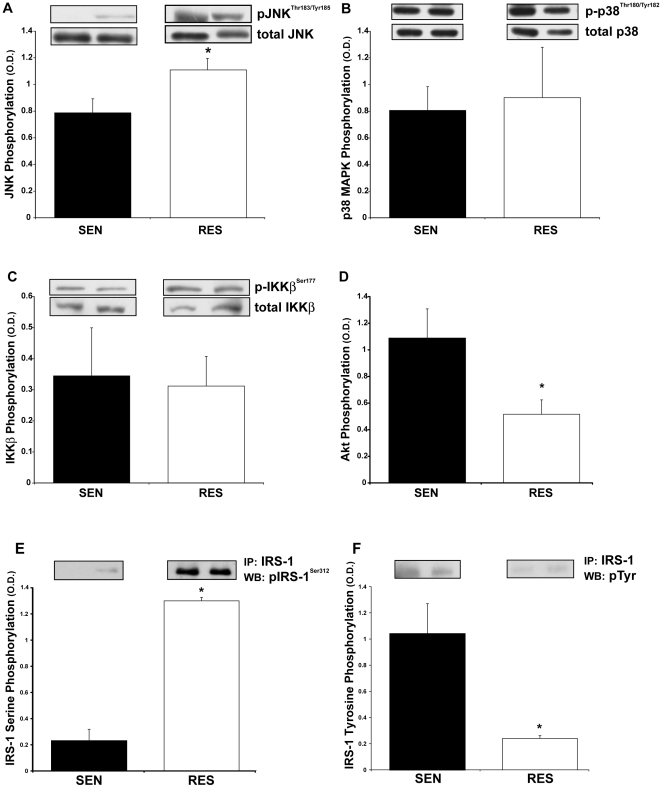
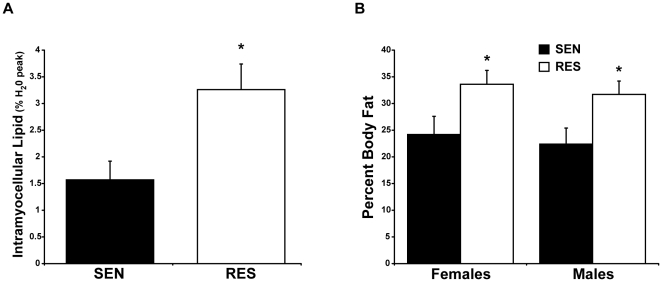
METHODOLOGY/PRINCIPAL FINDINGS: To explore whether specific stress kinases that have been implicated in the insulin resistance of obesity are potentially contributing to insulin resistance in non-obese individuals, twenty healthy, non-obese, normoglycemic subjects identified as insulin sensitive or resistant were studied. Vastus lateralis muscle biopsies obtained during euglycemic, hyperinsulinemic clamp were evaluated for insulin signaling and for activation of stress kinase pathways. Total and regional adipose stores and intramyocellular lipids (IMCL) were assessed by DXA, MRI and (1)H-MRS. In muscle of resistant subjects, phosphorylation of JNK was increased (1.36±0.23 vs. 0.78±0.10 OD units, P<0.05), while there was no evidence for activation of p38 MAPK or IKKβ. IRS-1 serine phosphorylation was increased (1.30±0.09 vs. 0.22±0.03 OD units, P<0.005) while insulin-stimulated tyrosine phosphorylation decreased (10.97±0.95 vs. 0.89±0.50 OD units, P<0.005). IMCL levels were twice as high in insulin resistant subjects (3.26±0.48 vs. 1.58±0.35% H(2)O peak, P<0.05), who also displayed increased total fat and abdominal fat when compared to insulin sensitive controls.
This is the first report demonstrating that insulin resistance in non-obese, normoglycemic subjects is associated with activation of the JNK pathway related to increased IMCL and higher total body and abdominal adipose stores. While JNK activation is consistent with a primary impact of muscle lipid accumulation on metabolic stress, further work is necessary to determine the relative contributions of the various mediators of impaired insulin signaling in this population.
在不肥胖的情况下,胰岛素抵抗的发病机制尚不清楚。在肥胖中,已经确定了多种应激激酶,它们通过关键第二信使蛋白的丝氨酸磷酸化来损害胰岛素信号通路。这些应激激酶通过与胰岛素靶组织和各种脂肪组织中局部脂质供应过多相关的各种机制而被激活。
方法/主要发现:为了探讨是否已经涉及肥胖症胰岛素抵抗的特定应激激酶可能导致非肥胖个体的胰岛素抵抗,研究了 20 名健康、非肥胖、血糖正常的个体,这些个体被确定为胰岛素敏感或抵抗。在正常血糖、高胰岛素血症钳夹期间从外侧股肌活检中评估胰岛素信号和应激激酶途径的激活。通过 DXA、MRI 和(1)H-MRS 评估总脂肪和局部脂肪储存和肌内甘油三酯(IMCL)。在抵抗组的肌肉中,JNK 的磷酸化增加(1.36±0.23 与 0.78±0.10 OD 单位,P<0.05),而 p38 MAPK 或 IKKβ 没有证据表明激活。IRS-1 丝氨酸磷酸化增加(1.30±0.09 与 0.22±0.03 OD 单位,P<0.005),而胰岛素刺激的酪氨酸磷酸化减少(10.97±0.95 与 0.89±0.50 OD 单位,P<0.005)。胰岛素抵抗组的 IMCL 水平是胰岛素敏感组的两倍(3.26±0.48 与 1.58±0.35% H2O 峰,P<0.05),与胰岛素敏感对照组相比,他们还显示出总脂肪和腹部脂肪增加。
这是第一项表明非肥胖、血糖正常的个体的胰岛素抵抗与 JNK 途径的激活相关的报告,该途径与增加的 IMCL 和更高的全身和腹部脂肪储存有关。虽然 JNK 激活与肌肉脂质积累对代谢应激的主要影响一致,但需要进一步的工作来确定在该人群中受损胰岛素信号的各种介质的相对贡献。