Leibniz Institute for Age Research - Fritz Lipmann Institute, Jena, Germany.
BMC Genomics. 2011 May 18;12(1):243. doi: 10.1186/1471-2164-12-243.
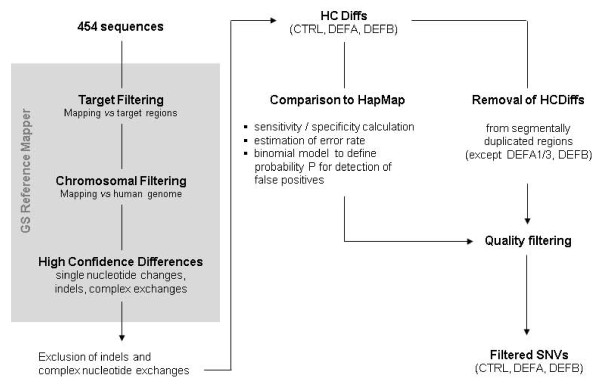
In highly copy number variable (CNV) regions such as the human defensin gene locus, comprehensive assessment of sequence variations is challenging. PCR approaches are practically restricted to tiny fractions, and next-generation sequencing (NGS) approaches of whole individual genomes e.g. by the 1000 Genomes Project is confined by an affordable sequence depth. Combining target enrichment with NGS may represent a feasible approach.
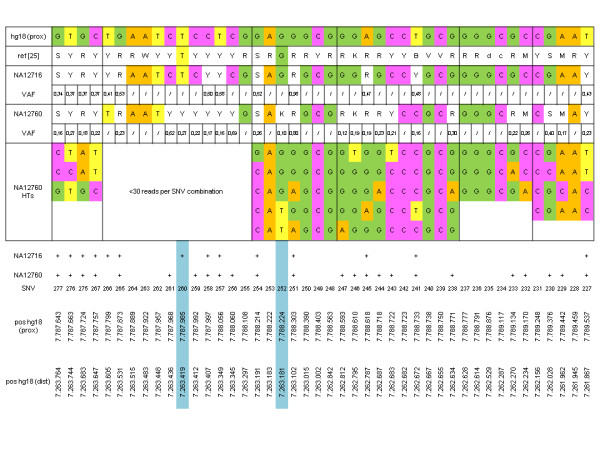
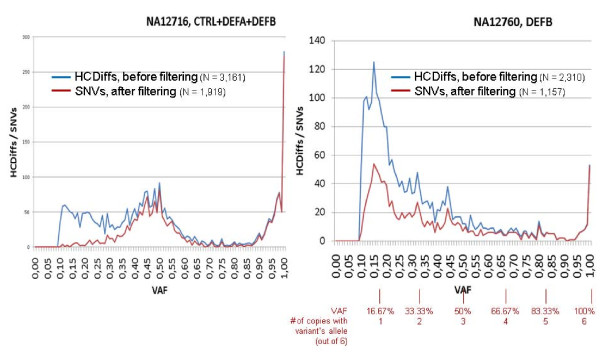
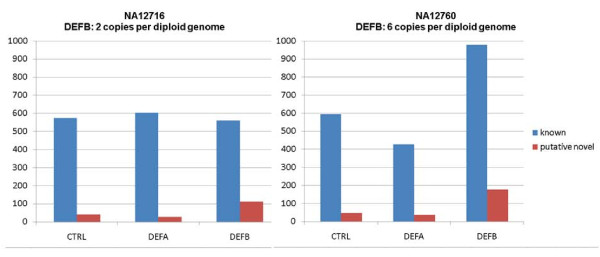
As a proof of principle, we enriched a ~850 kb section comprising the CNV defensin gene cluster DEFB, the invariable DEFA part and 11 control regions from two genomes by sequence capture and sequenced it by 454 technology. 6,651 differences to the human reference genome were found. Comparison to HapMap genotypes revealed sensitivities and specificities in the range of 94% to 99% for the identification of variations.Using error probabilities for rigorous filtering revealed 2,886 unique single nucleotide variations (SNVs) including 358 putative novel ones. DEFB CN determinations by haplotype ratios were in agreement with alternative methods.
Although currently labor extensive and having high costs, target enriched NGS provides a powerful tool for the comprehensive assessment of SNVs in highly polymorphic CNV regions of individual genomes. Furthermore, it reveals considerable amounts of putative novel variations and simultaneously allows CN estimation.
在高度拷贝数可变(CNV)区域,如人类防御素基因座,全面评估序列变异是具有挑战性的。PCR 方法实际上仅限于很小的部分,而下一代测序(NGS)方法,例如 1000 基因组计划,受限于可负担得起的测序深度。将靶向富集与 NGS 相结合可能是一种可行的方法。
作为原理验证,我们通过序列捕获对来自两个基因组的包含 CNV 防御素基因簇 DEFB、不变的 DEFA 部分和 11 个对照区域的约 850kb 区段进行了富集,并通过 454 技术进行了测序。与人类参考基因组相比,发现了 6651 个差异。与 HapMap 基因型的比较表明,用于识别变异的敏感性和特异性在 94%到 99%之间。使用错误概率进行严格过滤,揭示了 2886 个独特的单核苷酸变异(SNVs),包括 358 个可能的新变异。通过单倍型比的 DEFB CN 测定与替代方法一致。
尽管目前劳动强度大且成本高,但靶向富集 NGS 为个体基因组中高度多态性 CNV 区域的 SNV 全面评估提供了有力工具。此外,它揭示了大量可能的新变异,同时允许进行 CN 估计。