Institute of Bioinformatics, Johannes Kepler University, A-4040 Linz, Austria.
Nucleic Acids Res. 2012 May;40(9):e69. doi: 10.1093/nar/gks003. Epub 2012 Feb 1.
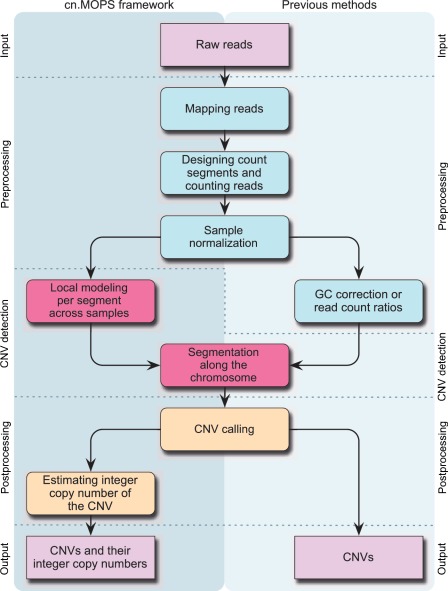
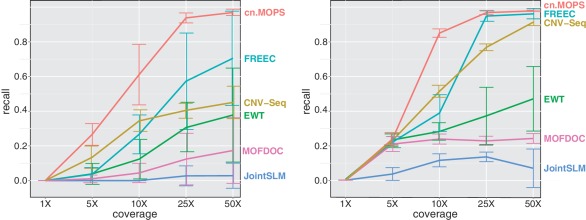
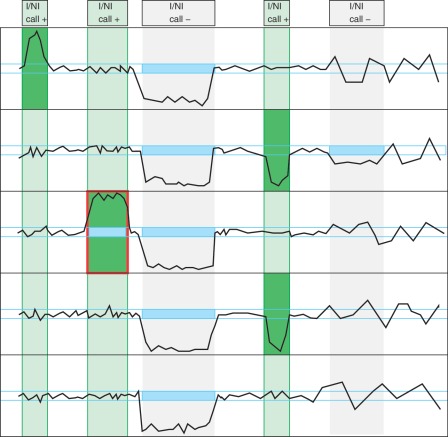
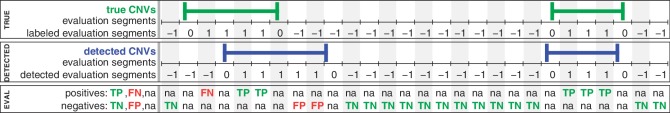
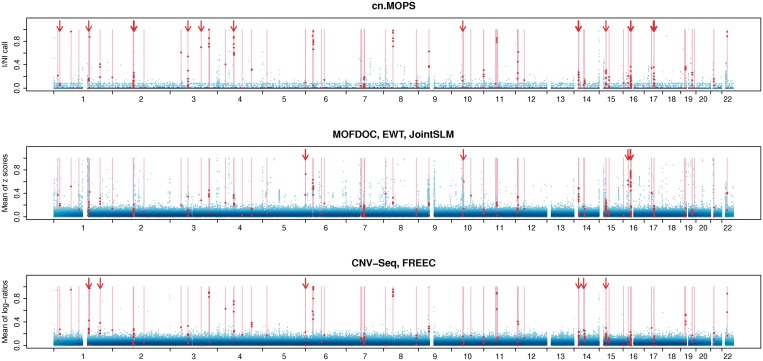
Quantitative analyses of next-generation sequencing (NGS) data, such as the detection of copy number variations (CNVs), remain challenging. Current methods detect CNVs as changes in the depth of coverage along chromosomes. Technological or genomic variations in the depth of coverage thus lead to a high false discovery rate (FDR), even upon correction for GC content. In the context of association studies between CNVs and disease, a high FDR means many false CNVs, thereby decreasing the discovery power of the study after correction for multiple testing. We propose 'Copy Number estimation by a Mixture Of PoissonS' (cn.MOPS), a data processing pipeline for CNV detection in NGS data. In contrast to previous approaches, cn.MOPS incorporates modeling of depths of coverage across samples at each genomic position. Therefore, cn.MOPS is not affected by read count variations along chromosomes. Using a Bayesian approach, cn.MOPS decomposes variations in the depth of coverage across samples into integer copy numbers and noise by means of its mixture components and Poisson distributions, respectively. The noise estimate allows for reducing the FDR by filtering out detections having high noise that are likely to be false detections. We compared cn.MOPS with the five most popular methods for CNV detection in NGS data using four benchmark datasets: (i) simulated data, (ii) NGS data from a male HapMap individual with implanted CNVs from the X chromosome, (iii) data from HapMap individuals with known CNVs, (iv) high coverage data from the 1000 Genomes Project. cn.MOPS outperformed its five competitors in terms of precision (1-FDR) and recall for both gains and losses in all benchmark data sets. The software cn.MOPS is publicly available as an R package at http://www.bioinf.jku.at/software/cnmops/ and at Bioconductor.
下一代测序(NGS)数据的定量分析,例如拷贝数变异(CNV)的检测,仍然具有挑战性。当前的方法通过检测染色体上覆盖深度的变化来检测 CNV。因此,即使对 GC 含量进行校正,覆盖深度的技术或基因组变异也会导致高假发现率(FDR)。在 CNV 与疾病之间的关联研究中,高 FDR 意味着许多假 CNV,从而在对多重检验进行校正后降低了研究的发现能力。我们提出了“通过泊松混合进行拷贝数估计(cn.MOPS)”,这是一种用于 NGS 数据中 CNV 检测的数据处理管道。与以前的方法不同,cn.MOPS 在每个基因组位置都整合了样本之间覆盖深度的建模。因此,cn.MOPS 不受染色体上读长变化的影响。通过使用贝叶斯方法,cn.MOPS 通过其混合成分和泊松分布将样本之间覆盖深度的变化分别分解为整数拷贝数和噪声。噪声估计通过过滤掉具有高噪声的检测来降低 FDR,这些检测很可能是假检测。我们使用四个基准数据集将 cn.MOPS 与用于 NGS 数据中 CNV 检测的五种最流行的方法进行了比较:(i)模拟数据,(ii)来自具有植入 X 染色体 CNV 的男性 HapMap 个体的 NGS 数据,(iii)具有已知 CNV 的 HapMap 个体的数据,(iv)来自 1000 基因组计划的高覆盖数据。cn.MOPS 在所有基准数据集的增益和损失方面均在精度(1-FDR)和召回率方面优于其五个竞争对手。cn.MOPS 软件作为 R 包在 http://www.bioinf.jku.at/software/cnmops/ 和 Bioconductor 上公开提供。