Division of Pharmaceutical Sciences, University of Wisconsin, Madison, WI, USA.
Neurobiol Dis. 2011 Sep;43(3):543-51. doi: 10.1016/j.nbd.2011.04.025. Epub 2011 May 6.
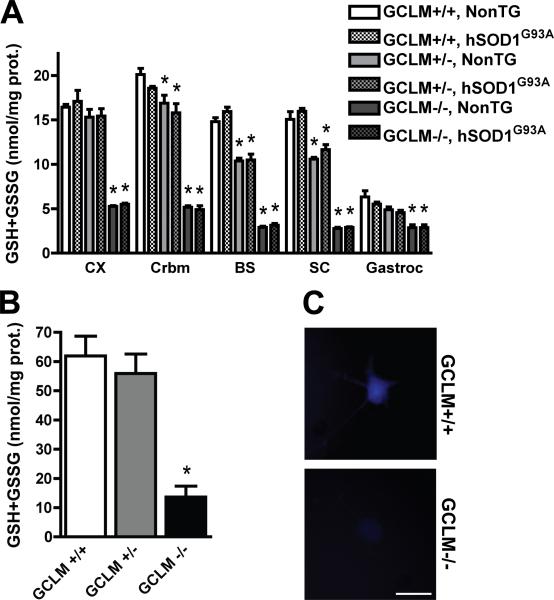
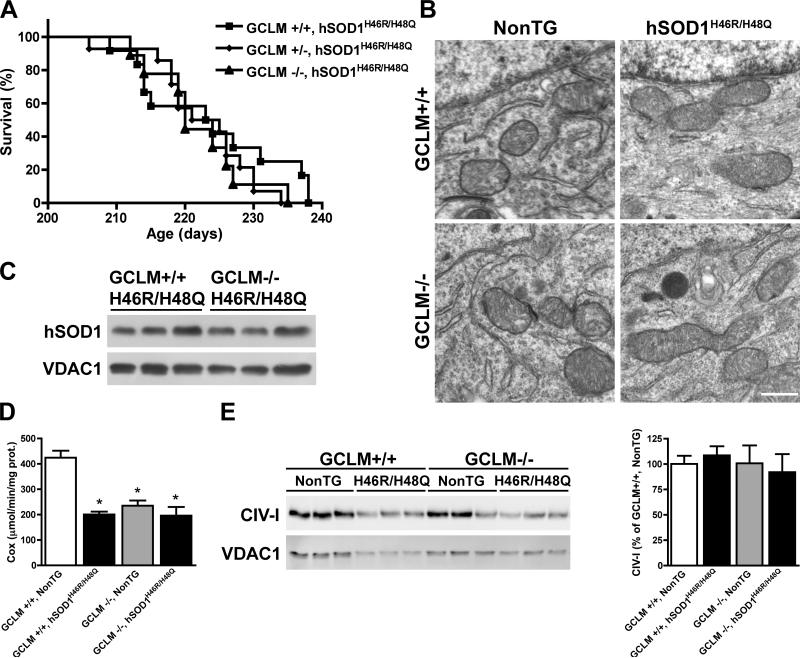
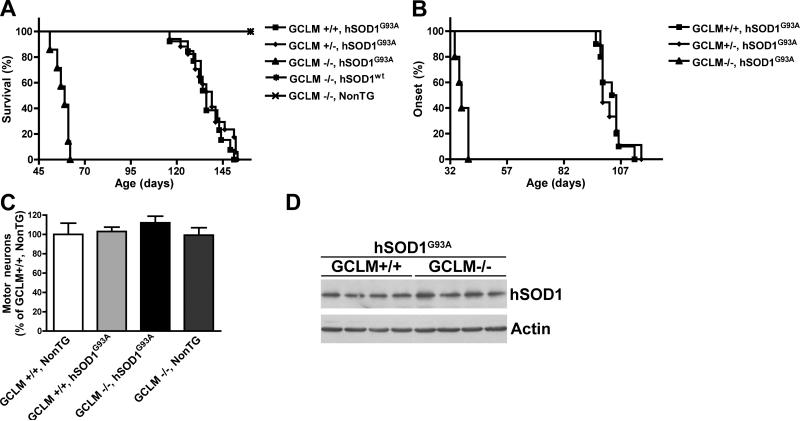
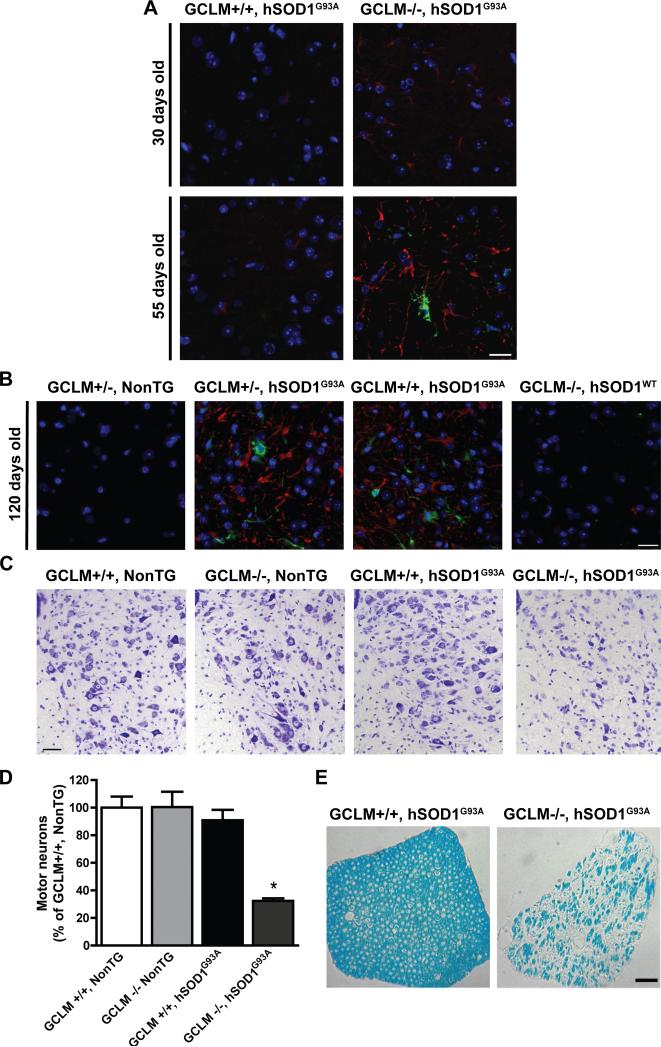
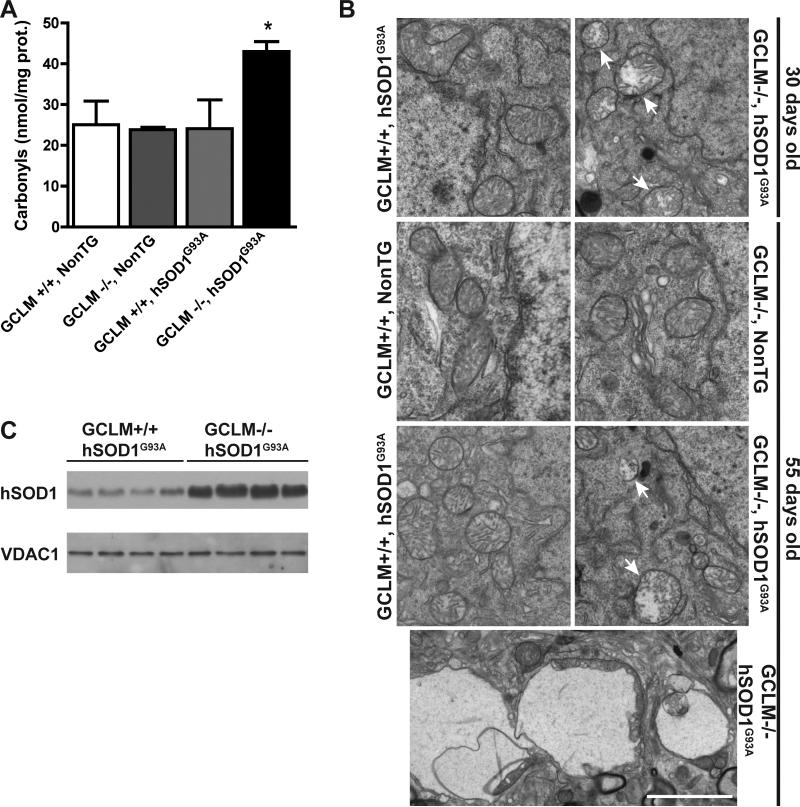
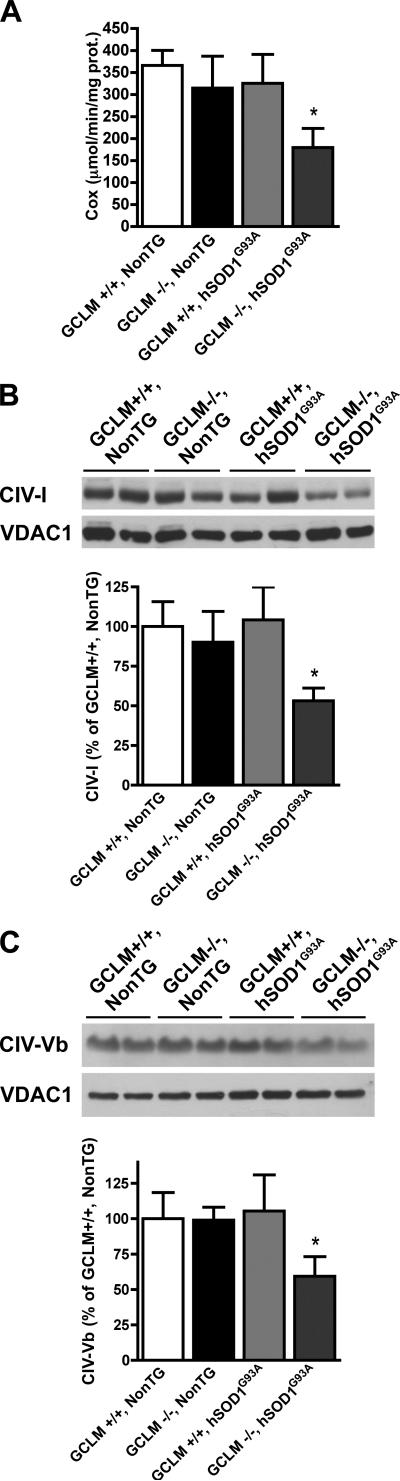
Dominant mutations in Cu/Zn-superoxide dismutase (SOD1) cause familial forms of amyotrophic lateral sclerosis (ALS), a fatal disorder characterized by the progressive loss of motor neurons. To investigate the role of antioxidant defenses in ALS we used knockout mice for the glutamate-cysteine ligase modifier subunit (GCLM-/-), which have a 70-80% reduction in total glutathione. Although GCLM(-/-) mice are viable and fertile, the life span of GCLM(-/-)/hSOD1(G93A) mice decreased in 55% when compared to GCLM(+/+)/hSOD1(G93A) mice. Decreased life span in GCLM(-/-)/hSOD1(G93A) mice was associated to increased oxidative stress, aggravated mitochondrial pathology and increased association of hSOD1 with the mitochondria. Interestingly, when the GCLM(-/-) animals were mated with a different ALS-model which overexpress the experimental mutation hSOD1(H46R/H48Q), no effect was observed in survival of GCLM(-/-)/hSOD1(H46R/H48Q) mice; and little or no mitochondrial pathology was observed. Since a specific disease modifier, such as glutathione deficiency, may affect only certain hSOD1 mutants, these findings contribute to our understanding of the potential difference in the molecular pathways by which different hSOD1 mutants generate disease.
铜/锌超氧化物歧化酶 (SOD1) 的显性突变导致家族性肌萎缩侧索硬化症 (ALS),这是一种致命的疾病,其特征是运动神经元的进行性丧失。为了研究抗氧化防御在 ALS 中的作用,我们使用了谷氨酸-半胱氨酸连接酶修饰亚基 (GCLM-/-) 的敲除小鼠,这些小鼠的总谷胱甘肽减少了 70-80%。尽管 GCLM(-/-) 小鼠具有活力和繁殖力,但与 GCLM(+/+)/hSOD1(G93A) 小鼠相比,GCLM(-/-)/hSOD1(G93A) 小鼠的寿命缩短了 55%。GCLM(-/-)/hSOD1(G93A) 小鼠寿命缩短与氧化应激增加、线粒体病理学加重以及 hSOD1 与线粒体的结合增加有关。有趣的是,当 GCLM(-/-) 动物与另一种表达实验性突变 hSOD1(H46R/H48Q) 的 ALS 模型交配时,GCLM(-/-)/hSOD1(H46R/H48Q) 小鼠的存活没有受到影响;并且观察到很少或没有线粒体病理学。由于特定的疾病修饰因子,如谷胱甘肽缺乏,可能仅影响某些 hSOD1 突变体,这些发现有助于我们理解不同 hSOD1 突变体产生疾病的分子途径的潜在差异。