Centre for Translational and Applied Genomics, BC Cancer Agency, Vancouver, British Columbia, Canada.
PLoS Comput Biol. 2011 May;7(5):e1001138. doi: 10.1371/journal.pcbi.1001138. Epub 2011 May 19.
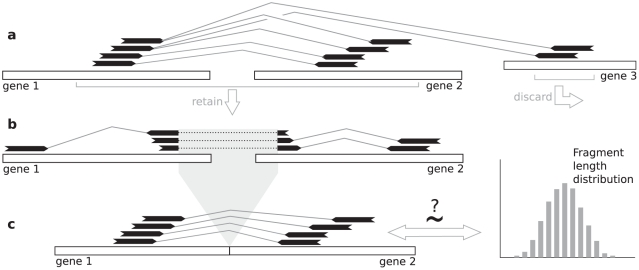
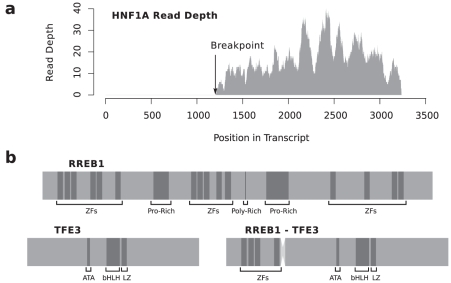
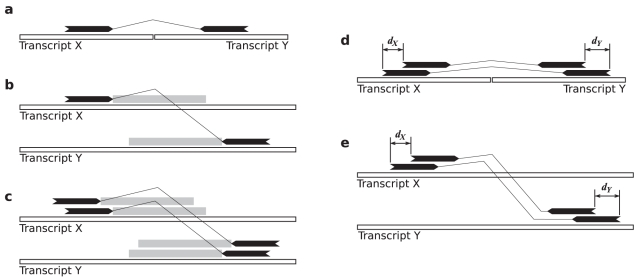
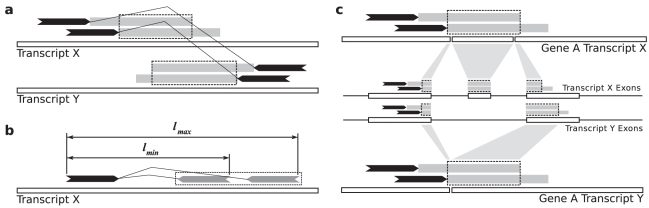
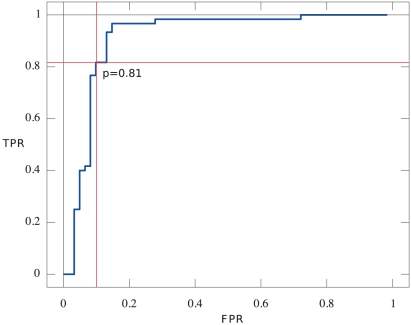
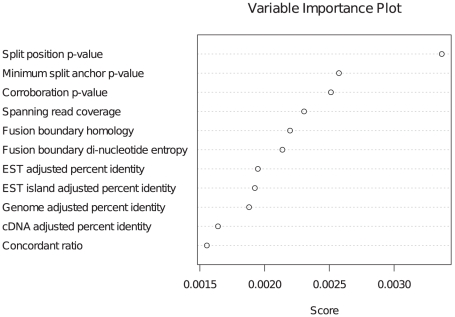
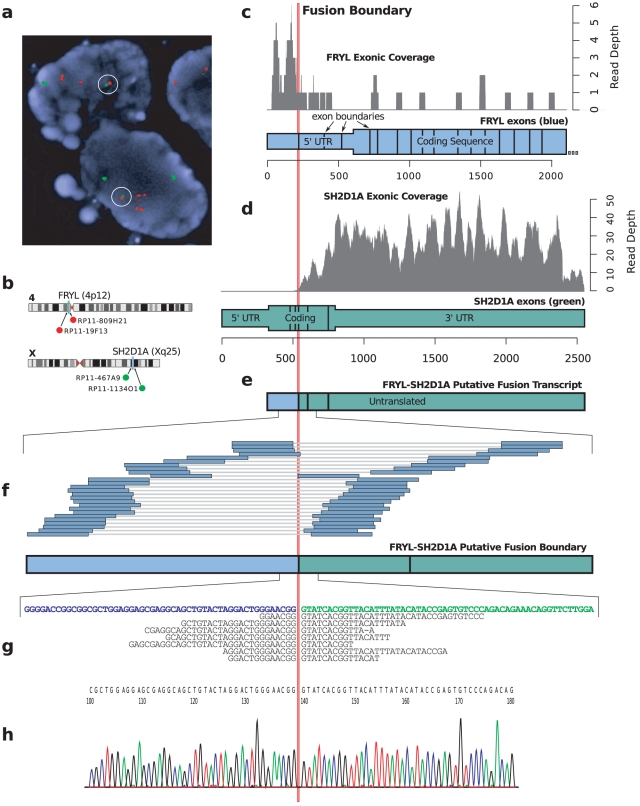
Gene fusions created by somatic genomic rearrangements are known to play an important role in the onset and development of some cancers, such as lymphomas and sarcomas. RNA-Seq (whole transcriptome shotgun sequencing) is proving to be a useful tool for the discovery of novel gene fusions in cancer transcriptomes. However, algorithmic methods for the discovery of gene fusions using RNA-Seq data remain underdeveloped. We have developed deFuse, a novel computational method for fusion discovery in tumor RNA-Seq data. Unlike existing methods that use only unique best-hit alignments and consider only fusion boundaries at the ends of known exons, deFuse considers all alignments and all possible locations for fusion boundaries. As a result, deFuse is able to identify fusion sequences with demonstrably better sensitivity than previous approaches. To increase the specificity of our approach, we curated a list of 60 true positive and 61 true negative fusion sequences (as confirmed by RT-PCR), and have trained an adaboost classifier on 11 novel features of the sequence data. The resulting classifier has an estimated value of 0.91 for the area under the ROC curve. We have used deFuse to discover gene fusions in 40 ovarian tumor samples, one ovarian cancer cell line, and three sarcoma samples. We report herein the first gene fusions discovered in ovarian cancer. We conclude that gene fusions are not infrequent events in ovarian cancer and that these events have the potential to substantially alter the expression patterns of the genes involved; gene fusions should therefore be considered in efforts to comprehensively characterize the mutational profiles of ovarian cancer transcriptomes.
体细胞基因组重排产生的基因融合已知在一些癌症(如淋巴瘤和肉瘤)的发生和发展中发挥重要作用。RNA-Seq(全转录组鸟枪法测序)被证明是发现癌症转录组中新型基因融合的有用工具。然而,使用 RNA-Seq 数据发现基因融合的算法方法仍未得到充分发展。我们开发了 deFuse,这是一种用于肿瘤 RNA-Seq 数据中融合发现的新型计算方法。与仅使用唯一最佳命中比对并仅考虑已知外显子末端融合边界的现有方法不同,deFuse 考虑了所有比对和融合边界的所有可能位置。因此,deFuse 能够以明显优于先前方法的灵敏度识别融合序列。为了提高我们方法的特异性,我们精心挑选了 60 个真阳性和 61 个真阴性融合序列(通过 RT-PCR 确认),并在序列数据的 11 个新特征上训练了 adaboost 分类器。由此产生的分类器在 ROC 曲线下的估计值为 0.91。我们使用 deFuse 在 40 个卵巢肿瘤样本、一个卵巢癌细胞系和三个肉瘤样本中发现了基因融合。本文报告了在卵巢癌中发现的第一个基因融合。我们得出的结论是,基因融合在卵巢癌中并非罕见事件,这些事件有可能极大地改变所涉及基因的表达模式;因此,在努力全面描述卵巢癌转录组的突变谱时,应考虑基因融合。