Department of Chemical Physiology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037, USA.
J Proteome Res. 2011 Aug 5;10(8):3690-700. doi: 10.1021/pr200304u. Epub 2011 Jun 24.
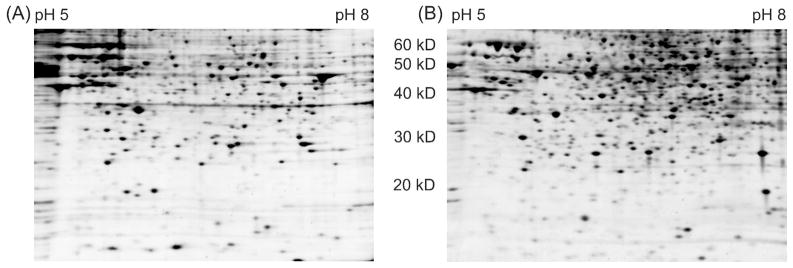
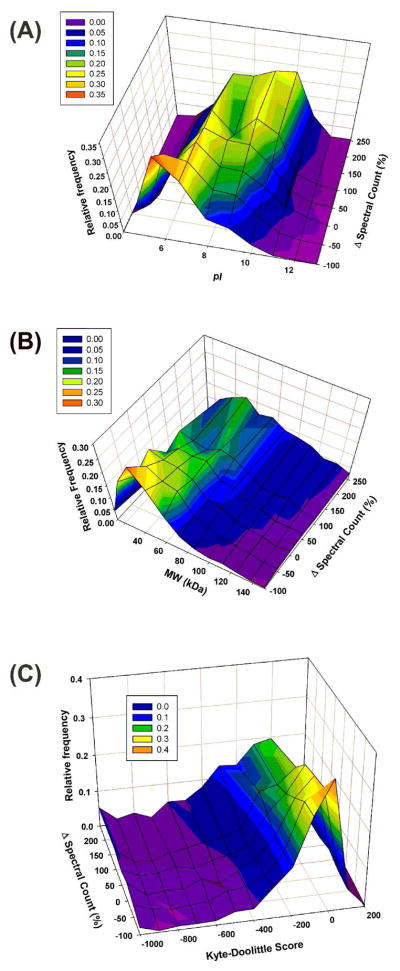
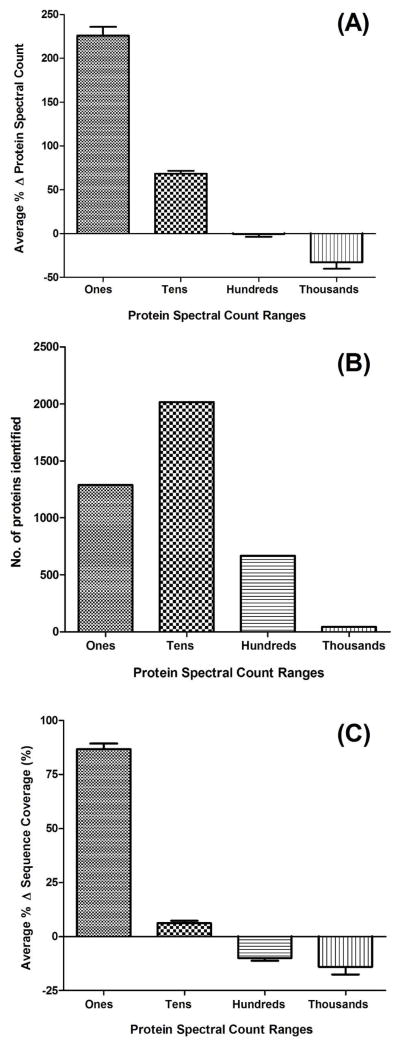
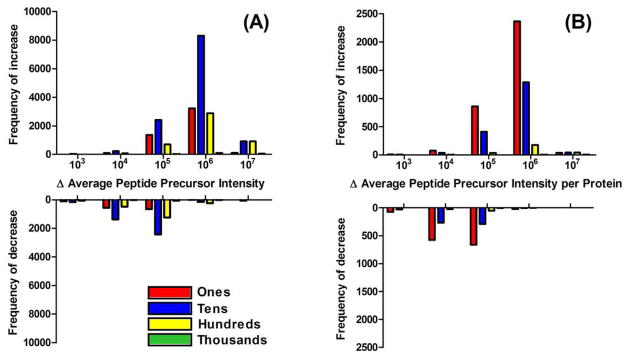
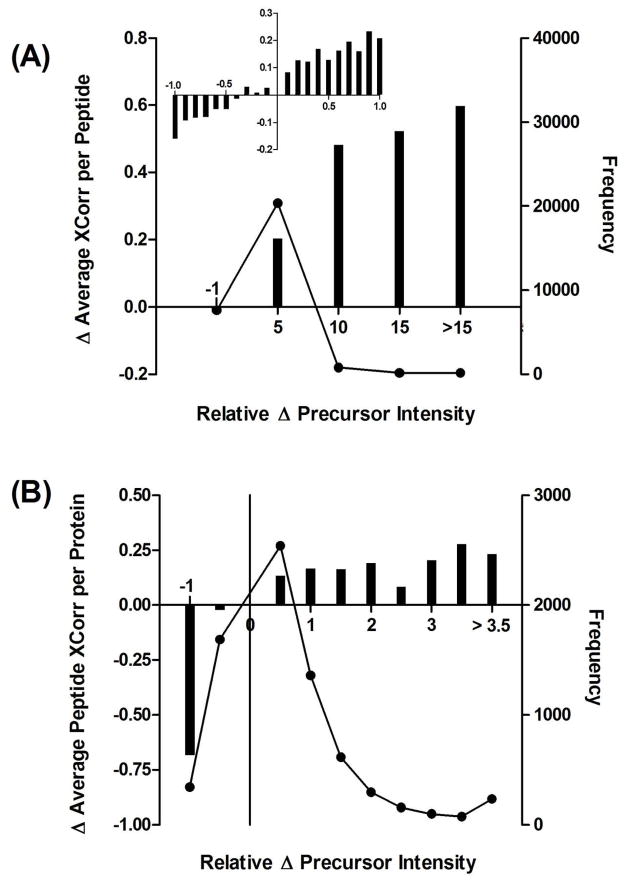
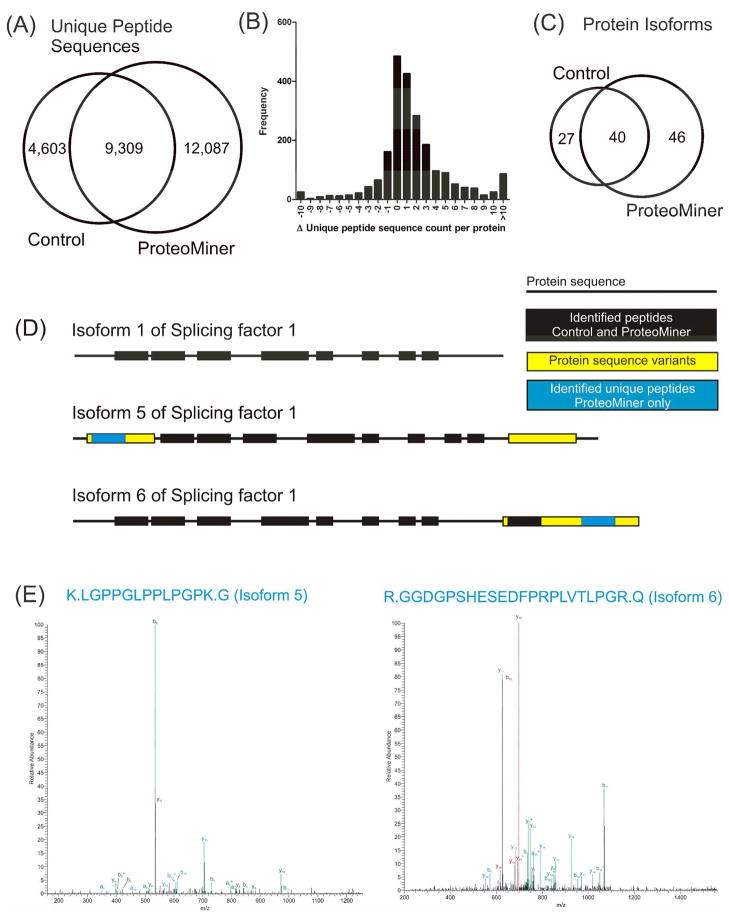
Ideally, shotgun proteomics would facilitate the identification of an entire proteome with 100% protein sequence coverage. In reality, the large dynamic range and complexity of cellular proteomes results in oversampling of abundant proteins, while peptides from low abundance proteins are undersampled or remain undetected. We tested the proteome equalization technology, ProteoMiner, in conjunction with Multidimensional Protein Identification Technology (MudPIT) to determine how the equalization of protein dynamic range could improve shotgun proteomics methods for the analysis of cellular proteomes. Our results suggest low abundance protein identifications were improved by two mechanisms: (1) depletion of high abundance proteins freed ion trap sampling space usually occupied by high abundance peptides and (2) enrichment of low abundance proteins increased the probability of sampling their corresponding more abundant peptides. Both mechanisms also contributed to dramatic increases in the quantity of peptides identified and the quality of MS/MS spectra acquired due to increases in precursor intensity of peptides from low abundance proteins. From our large data set of identified proteins, we categorized the dominant physicochemical factors that facilitate proteome equalization with a hexapeptide library. These results illustrate that equalization of the dynamic range of the cellular proteome is a promising methodology to improve low abundance protein identification confidence, reproducibility, and sequence coverage in shotgun proteomics experiments, opening a new avenue of research for improving proteome coverage.
理想情况下,鸟枪法蛋白质组学可以实现对整个蛋白质组的 100%序列覆盖鉴定。但实际上,由于细胞蛋白质组具有较大的动态范围和复杂性,大量存在的蛋白质会被过度采样,而低丰度蛋白质的肽段则被采样不足或未被检测到。我们联合使用多维蛋白质鉴定技术(MudPIT)和 ProteoMiner 蛋白质组均一化技术来检测蛋白质组均一化技术,以确定蛋白质动态范围的均一化如何改善用于分析细胞蛋白质组的鸟枪法蛋白质组学方法。我们的研究结果表明,有两种机制可以改善低丰度蛋白质的鉴定:(1)耗尽高丰度蛋白质可以释放离子阱采样空间,通常这些空间被高丰度肽占据;(2)低丰度蛋白质的富集增加了采样其相应更丰富肽的概率。这两种机制也有助于由于低丰度蛋白质的肽的前体强度增加,从而显著增加了鉴定的肽数量和获得的 MS/MS 谱的质量。根据我们的大量鉴定蛋白质数据集,我们使用六肽文库对促进蛋白质组均一化的主要物理化学因素进行了分类。这些结果表明,细胞蛋白质组的动态范围均一化是一种很有前途的方法,可以提高鸟枪法蛋白质组学实验中低丰度蛋白质鉴定的置信度、重现性和序列覆盖率,为提高蛋白质组覆盖率开辟了新的研究途径。