Department of Clinical and Experimental Epilepsy, UCL Institute of Neurology and National Hospital for Neurology and Neurosurgery, UCL, Queen Square, London WC1N 3BG, UK.
Brain. 2011 Oct;134(Pt 10):2982-3010. doi: 10.1093/brain/awr129. Epub 2011 Jun 29.
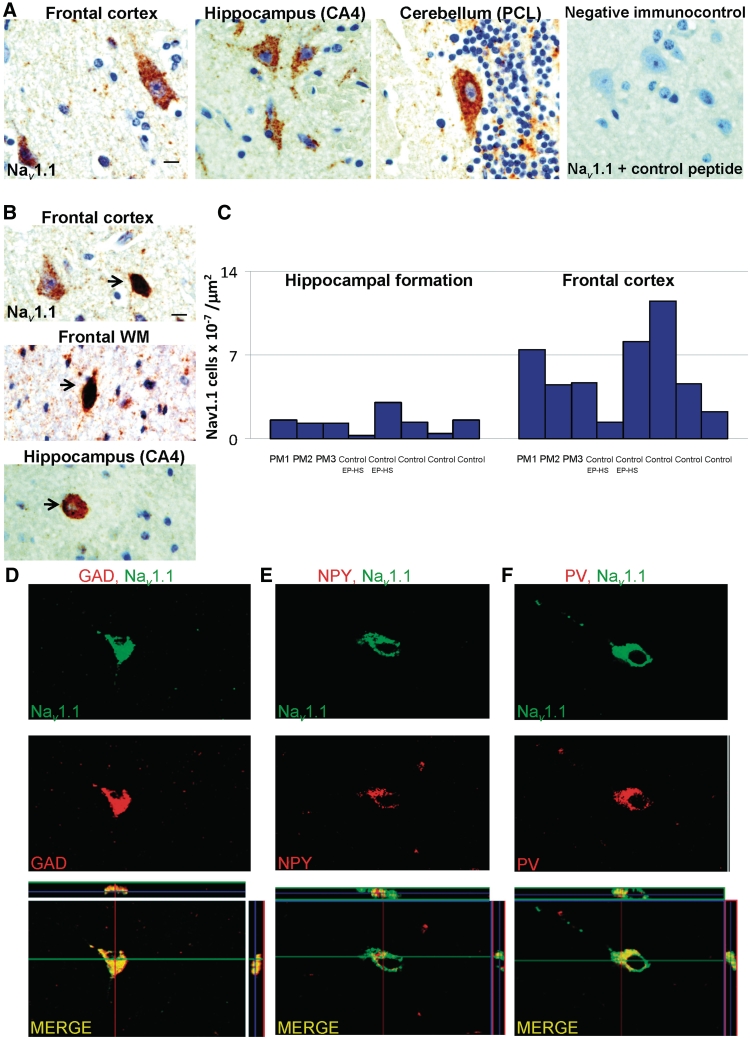
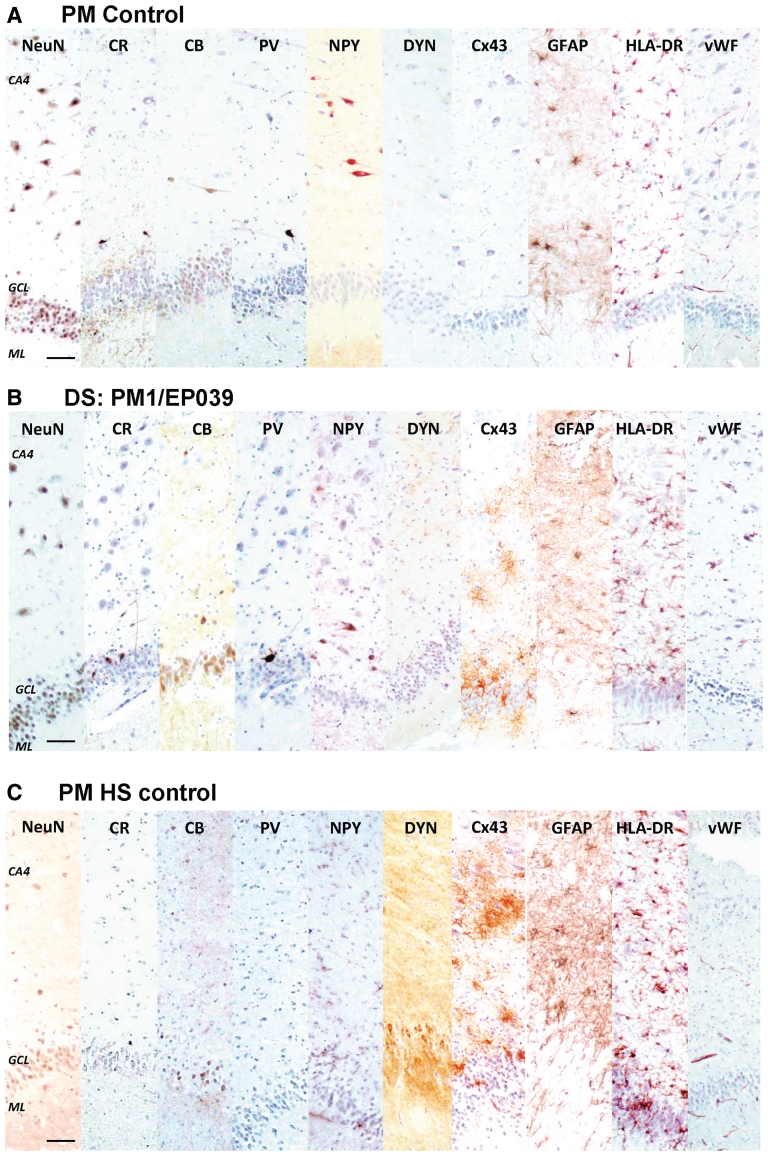
Dravet syndrome is an epilepsy syndrome of infantile onset, frequently caused by SCN1A mutations or deletions. Its prevalence, long-term evolution in adults and neuropathology are not well known. We identified a series of 22 adult patients, including three adult post-mortem cases with Dravet syndrome. For all patients, we reviewed the clinical history, seizure types and frequency, antiepileptic drugs, cognitive, social and functional outcome and results of investigations. A systematic neuropathology study was performed, with post-mortem material from three adult cases with Dravet syndrome, in comparison with controls and a range of relevant paediatric tissue. Twenty-two adults with Dravet syndrome, 10 female, were included, median age 39 years (range 20-66). SCN1A structural variation was found in 60% of the adult Dravet patients tested, including one post-mortem case with DNA extracted from brain tissue. Novel mutations were described for 11 adult patients; one patient had three SCN1A mutations. Features of Dravet syndrome in adulthood include multiple seizure types despite polytherapy, and age-dependent evolution in seizure semiology and electroencephalographic pattern. Fever sensitivity persisted through adulthood in 11 cases. Neurological decline occurred in adulthood with cognitive and motor deterioration. Dysphagia may develop in or after the fourth decade of life, leading to significant morbidity, or death. The correct diagnosis at an older age made an impact at several levels. Treatment changes improved seizure control even after years of drug resistance in all three cases with sufficient follow-up after drug changes were instituted; better control led to significant improvement in cognitive performance and quality of life in adulthood in two cases. There was no histopathological hallmark feature of Dravet syndrome in this series. Strikingly, there was remarkable preservation of neurons and interneurons in the neocortex and hippocampi of Dravet adult post-mortem cases. Our study provides evidence that Dravet syndrome is at least in part an epileptic encephalopathy.
德拉维雷特综合征是一种婴儿期起病的癫痫综合征,常由 SCN1A 突变或缺失引起。其患病率、成人长期演变和神经病理学特征尚不清楚。我们鉴定了一系列 22 例成年患者,其中包括 3 例成年尸检德拉维雷特综合征病例。对所有患者,我们均回顾了临床病史、发作类型和频率、抗癫痫药物、认知、社会和功能结局以及检查结果。对 3 例成年德拉维雷特综合征尸检病例,与对照组及一系列相关儿科组织进行了系统神经病理学研究。纳入了 22 例成年德拉维雷特综合征患者,其中 10 例为女性,中位年龄 39 岁(范围 20-66 岁)。在接受测试的成年德拉维雷特综合征患者中,60%发现 SCN1A 结构变异,包括 1 例从脑组织提取 DNA 的尸检病例。描述了 11 例成年患者的新突变;1 例患者有 3 个 SCN1A 突变。成年期德拉维雷特综合征的特征包括尽管进行了多药治疗,但仍有多种发作类型,且发作的症状学和脑电图模式随年龄而变化。11 例病例在成年期仍有发热敏感性。认知和运动功能恶化导致成年期神经功能下降。吞咽困难可能在 40 岁后或 40 岁后出现,导致显著的发病率或死亡。在较晚的年龄做出正确诊断在多个层面产生了影响。在所有 3 例有足够药物改变后随访的病例中,治疗改变改善了即使在药物耐药多年后的发作控制;在成年期更好的控制导致认知表现和生活质量显著改善。本系列研究中没有发现德拉维雷特综合征的组织病理学特征。引人注目的是,在成年尸检德拉维雷特综合征病例的新皮质和海马区,神经元和中间神经元明显保存。我们的研究提供了证据表明,德拉维雷特综合征至少部分是一种癫痫性脑病。