Laboratory for Prenatal Diagnostics of Inherited Diseases, Ott's Institute of Obstetrics and Gynecology RAMS, Mendeleevskaya line 3, Saint-Petersburg, Russia.
BMC Med Genet. 2011 Jul 15;12:96. doi: 10.1186/1471-2350-12-96.
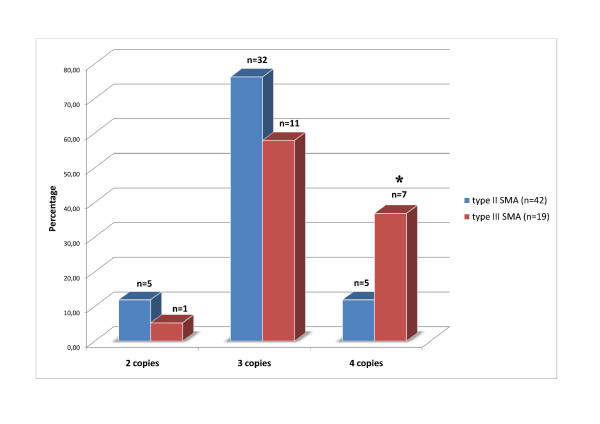
Spinal muscular atrophy (SMA type I, II and III) is an autosomal recessive neuromuscular disorder caused by mutations in the survival motor neuron gene (SMN1). SMN2 is a centromeric copy gene that has been characterized as a major modifier of SMA severity. SMA type I patients have one or two SMN2 copies while most SMA type II patients carry three SMN2 copies and SMA III patients have three or four SMN2 copies. The SMN1 gene produces a full-length transcript (FL-SMN) while SMN2 is only able to produce a small portion of the FL-SMN because of a splice mutation which results in the production of abnormal SMNΔ7 mRNA.
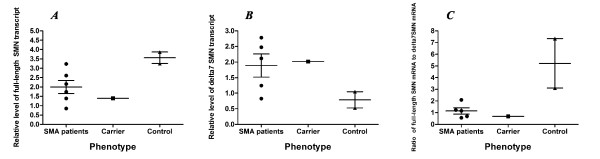
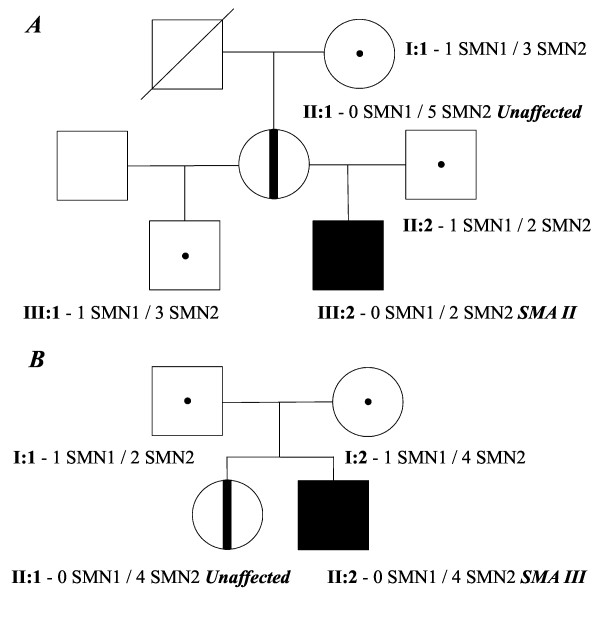
In this study we performed quantification of the SMN2 gene copy number in Russian patients affected by SMA type II and III (42 and 19 patients, respectively) by means of real-time PCR. Moreover, we present two families consisting of asymptomatic carriers of a homozygous absence of the SMN1 gene. We also developed a novel RT-qPCR-based assay to determine the FL-SMN/SMNΔ7 mRNA ratio as SMA biomarker.
Comparison of the SMN2 copy number and clinical features revealed a significant correlation between mild clinical phenotype (SMA type III) and presence of four copies of the SMN2 gene. In both asymptomatic cases we found an increased number of SMN2 copies in the healthy carriers and a biallelic SMN1 absence. Furthermore, the novel assay revealed a difference between SMA patients and healthy controls.
We suggest that the SMN2 gene copy quantification in SMA patients could be used as a prognostic tool for discrimination between the SMA type II and SMA type III diagnoses, whereas the FL-SMN/SMNΔ7 mRNA ratio could be a useful biomarker for detecting changes during SMA pharmacotherapy.
脊髓性肌萎缩症(SMA 型 I、II 和 III)是一种常染色体隐性神经肌肉疾病,由生存运动神经元基因(SMN1)的突变引起。SMN2 是一个着丝粒拷贝基因,已被确定为 SMA 严重程度的主要修饰基因。SMA 型 I 患者有一个或两个 SMN2 拷贝,而大多数 SMA 型 II 患者携带三个 SMN2 拷贝,SMA 型 III 患者携带三个或四个 SMN2 拷贝。SMN1 基因产生全长转录本(FL-SMN),而 SMN2 由于剪接突变只能产生 FL-SMN 的一小部分,导致异常 SMNΔ7 mRNA 的产生。
本研究通过实时 PCR 对俄罗斯 SMA 型 II 和 III 患者(分别为 42 名和 19 名患者)的 SMN2 基因拷贝数进行定量。此外,我们介绍了两个由 SMN1 基因纯合缺失的无症状携带者组成的家族。我们还开发了一种新的基于 RT-qPCR 的测定法来确定 FL-SMN/SMNΔ7 mRNA 比值作为 SMA 生物标志物。
SMN2 拷贝数与临床特征的比较表明,轻度临床表型(SMA 型 III)与存在四个 SMN2 基因拷贝之间存在显著相关性。在两个无症状病例中,我们发现健康携带者的 SMN2 拷贝数增加,并且存在 SMN1 基因的双等位基因缺失。此外,新的测定法显示 SMA 患者与健康对照之间存在差异。
我们建议,在 SMA 患者中,SMN2 基因拷贝数的定量可作为区分 SMA 型 II 和 SMA 型 III 诊断的预后工具,而 FL-SMN/SMNΔ7 mRNA 比值可作为检测 SMA 药物治疗期间变化的有用生物标志物。