INRA, UMR1202 BIOGECO, F-33610 Cestas, France.
BMC Genomics. 2011 Jul 18;12:368. doi: 10.1186/1471-2164-12-368.
Single nucleotide polymorphisms (SNPs) are the most abundant source of genetic variation among individuals of a species. New genotyping technologies allow examining hundreds to thousands of SNPs in a single reaction for a wide range of applications such as genetic diversity analysis, linkage mapping, fine QTL mapping, association studies, marker-assisted or genome-wide selection. In this paper, we evaluated the potential of highly-multiplexed SNP genotyping for genetic mapping in maritime pine (Pinus pinaster Ait.), the main conifer used for commercial plantation in southwestern Europe.
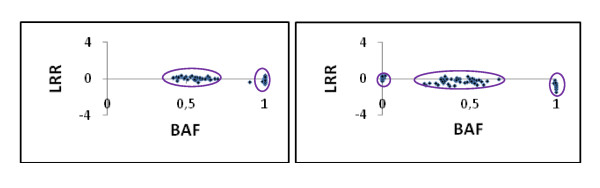
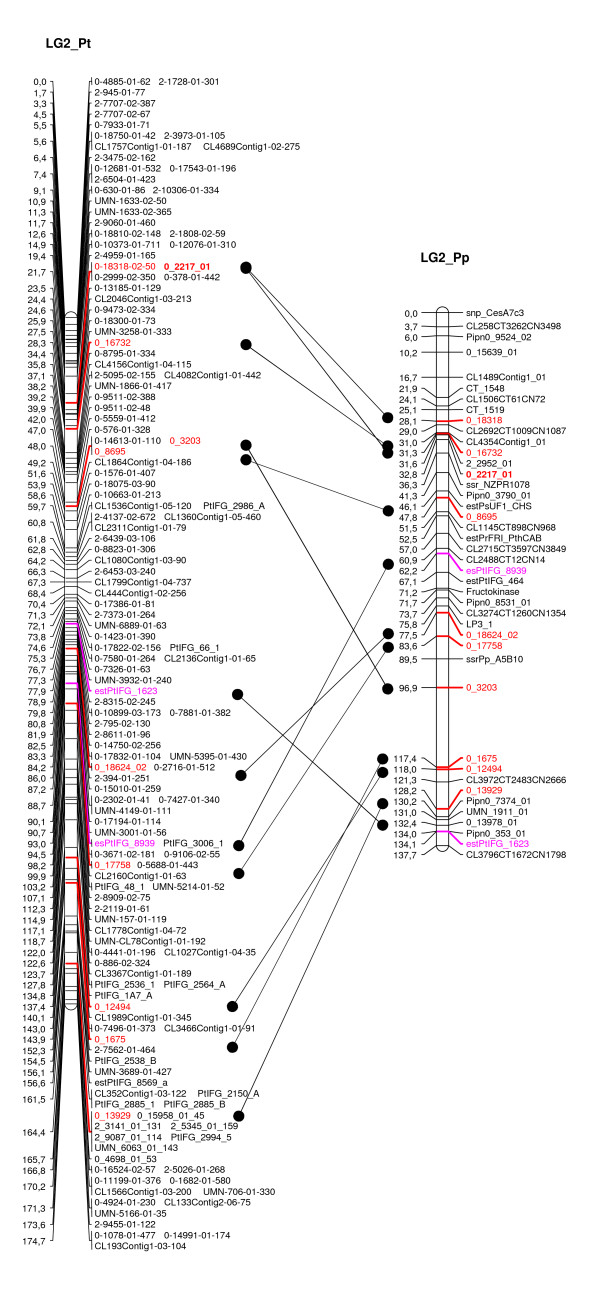
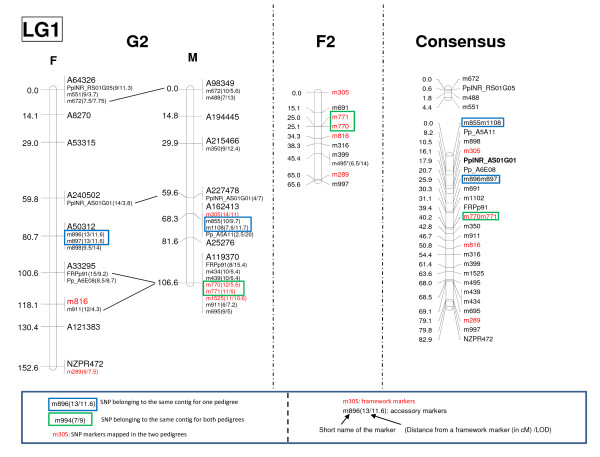
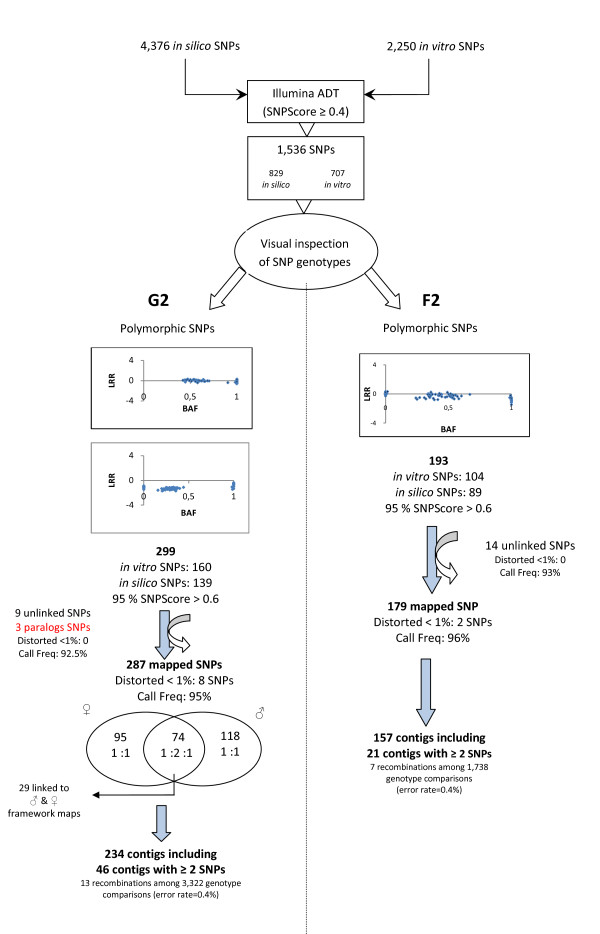
We designed a custom GoldenGate assay for 1,536 SNPs detected through the resequencing of gene fragments (707 in vitro SNPs/Indels) and from Sanger-derived Expressed Sequenced Tags assembled into a unigene set (829 in silico SNPs/Indels). Offspring from three-generation outbred (G2) and inbred (F2) pedigrees were genotyped. The success rate of the assay was 63.6% and 74.8% for in silico and in vitro SNPs, respectively. A genotyping error rate of 0.4% was further estimated from segregating data of SNPs belonging to the same gene. Overall, 394 SNPs were available for mapping. A total of 287 SNPs were integrated with previously mapped markers in the G2 parental maps, while 179 SNPs were localized on the map generated from the analysis of the F2 progeny. Based on 98 markers segregating in both pedigrees, we were able to generate a consensus map comprising 357 SNPs from 292 different loci. Finally, the analysis of sequence homology between mapped markers and their orthologs in a Pinus taeda linkage map, made it possible to align the 12 linkage groups of both species.
Our results show that the GoldenGate assay can be used successfully for high-throughput SNP genotyping in maritime pine, a conifer species that has a genome seven times the size of the human genome. This SNP-array will be extended thanks to recent sequencing effort using new generation sequencing technologies and will include SNPs from comparative orthologous sequences that were identified in the present study, providing a wider collection of anchor points for comparative genomics among the conifers.
单核苷酸多态性(SNP)是物种个体间遗传变异的最丰富来源。新的基因分型技术允许在单个反应中检查数百到数千个 SNP,适用于广泛的应用,如遗传多样性分析、连锁作图、精细 QTL 作图、关联研究、标记辅助或全基因组选择。在本文中,我们评估了高度多重 SNP 基因分型在海洋松(Pinus pinaster Ait.)遗传作图中的潜力,海洋松是欧洲西南部商业种植的主要针叶树。
我们设计了一种定制的 GoldenGate 测定法,用于检测通过基因片段重测序(707 个体外 SNP/Indels)和来自桑格衍生的表达序列标签组装成一个基因集(829 个计算机 SNP/Indels)检测到的 1536 个 SNP。三代杂交(G2)和近交(F2)系谱的后代进行了基因分型。体外和计算机 SNP 的测定成功率分别为 63.6%和 74.8%。从属于同一基因的 SNP 的分离数据中进一步估计了基因分型错误率为 0.4%。总体而言,有 394 个 SNP 可用于作图。共有 287 个 SNP 整合到 G2 亲本图谱中的先前映射标记中,而 179 个 SNP 定位在来自 F2 后代分析的图谱上。基于在两个系谱中分离的 98 个标记,我们能够生成一个包含 357 个 SNP 的共识图谱,这些 SNP 来自 292 个不同的基因座。最后,对映射标记与其在火炬松连锁图谱中的同源物之间的序列同源性进行分析,使得可以对两个物种的 12 个连锁群进行对齐。
我们的结果表明,GoldenGate 测定法可成功用于海洋松的高通量 SNP 基因分型,海洋松的基因组大小是人类基因组的七倍。由于最近使用新一代测序技术进行测序的努力,该 SNP 芯片将得到扩展,并将包括在本研究中鉴定的比较同源序列的 SNP,为针叶树之间的比较基因组学提供更广泛的锚点集合。