Bartholomé Jérôme, Bink Marco Cam, van Heerwaarden Joost, Chancerel Emilie, Boury Christophe, Lesur Isabelle, Isik Fikret, Bouffier Laurent, Plomion Christophe
BIOGECO, INRA, Univ. Bordeaux, 33610, Cestas, France.
Biometris, Wageningen University and Research Centre, NL-6700 AC, Wageningen, Netherlands.
PLoS One. 2016 Nov 2;11(11):e0165323. doi: 10.1371/journal.pone.0165323. eCollection 2016.
Increasing our understanding of the genetic architecture of complex traits, through analyses of genotype-phenotype associations and of the genes/polymorphisms accounting for trait variation, is crucial, to improve the integration of molecular markers into forest tree breeding. In this study, two full-sib families and one breeding population of maritime pine were used to identify quantitative trait loci (QTLs) for height growth and stem straightness, through linkage analysis (LA) and linkage disequilibrium (LD) mapping approaches.
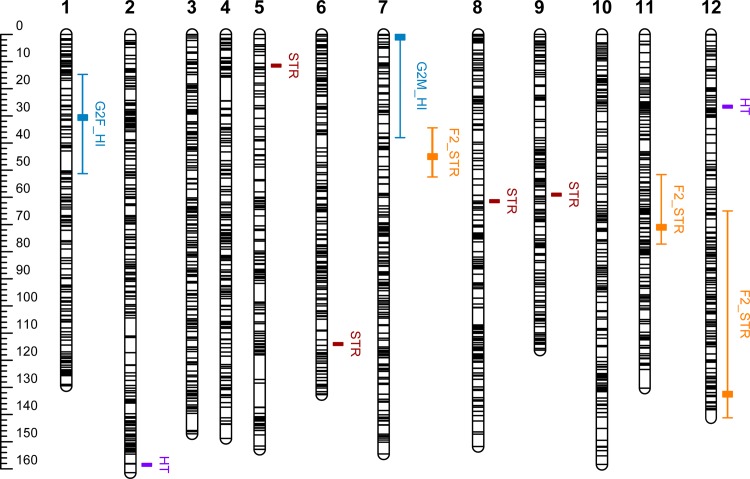
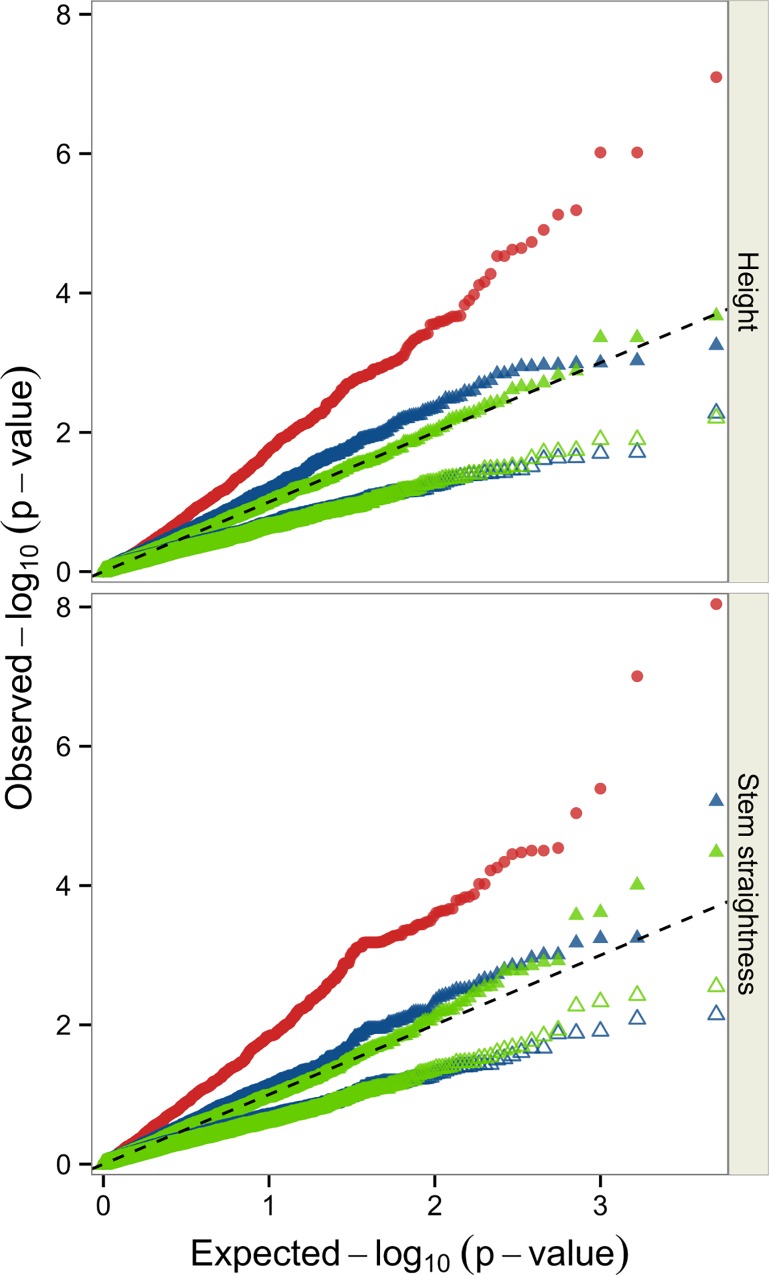
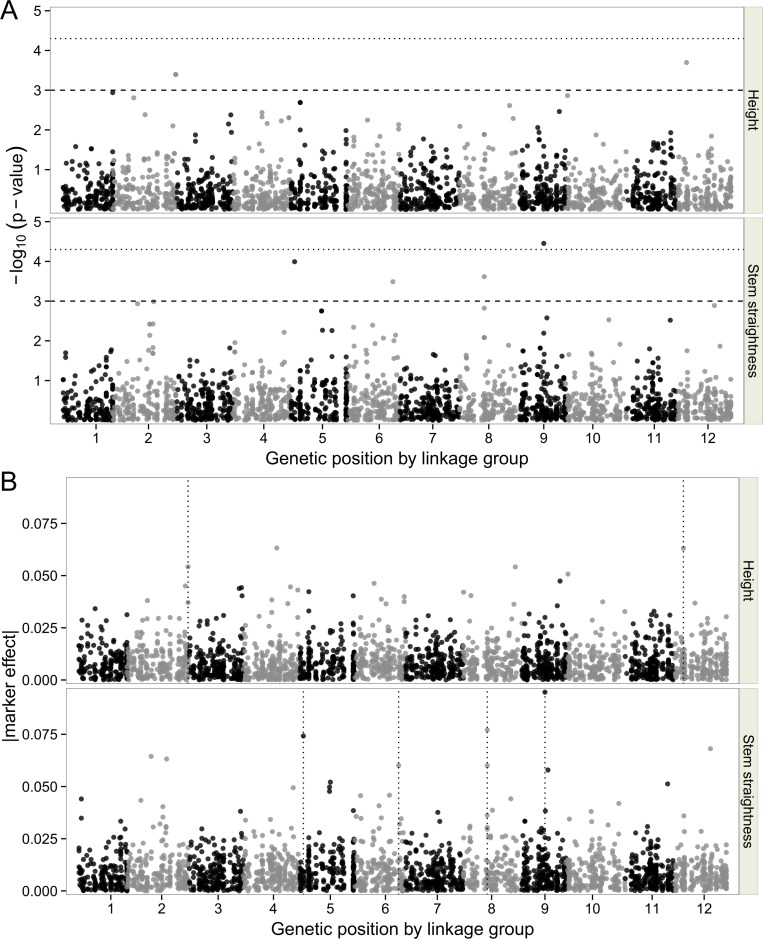
The populations used for LA consisted of two unrelated three-generation full-sib families (n = 197 and n = 477). These populations were assessed for height growth or stem straightness and genotyped for 248 and 217 markers, respectively. The population used for LD mapping consisted of 661 founders of the first and second generations of the breeding program. This population was phenotyped for the same traits and genotyped for 2,498 single-nucleotide polymorphism (SNP) markers corresponding to 1,652 gene loci. The gene-based reference genetic map of maritime pine was used to localize and compare the QTLs detected by the two approaches, for both traits. LA identified three QTLs for stem straightness and two QTLs for height growth. The LD study yielded seven significant associations (P ≤ 0.001): four for stem straightness and three for height growth. No colocalisation was found between QTLs identified by LA and SNPs detected by LD mapping for the same trait.
This study provides the first comparison of LA and LD mapping approaches in maritime pine, highlighting the complementary nature of these two approaches for deciphering the genetic architecture of two mandatory traits of the breeding program.
通过分析基因型与表型的关联以及解释性状变异的基因/多态性,加深我们对复杂性状遗传结构的理解,对于改善分子标记在林木育种中的整合至关重要。在本研究中,利用两个全同胞家系和一个欧洲黑松育种群体,通过连锁分析(LA)和连锁不平衡(LD)作图方法,鉴定了与树高生长和树干通直度相关的数量性状位点(QTL)。
用于LA的群体由两个不相关的三代全同胞家系组成(n = 197和n = 477)。对这些群体进行了树高生长或树干通直度评估,并分别对248个和217个标记进行了基因分型。用于LD作图的群体由育种计划第一代和第二代的661个亲本组成。对该群体进行了相同性状的表型分析,并对对应于1652个基因座的2498个单核苷酸多态性(SNP)标记进行了基因分型。利用基于基因的欧洲黑松参考遗传图谱对两种方法检测到的两个性状的QTL进行定位和比较。LA鉴定出3个与树干通直度相关的QTL和2个与树高生长相关的QTL。LD研究产生了7个显著关联(P≤0.001):4个与树干通直度相关,3个与树高生长相关。对于同一性状,LA鉴定的QTL与LD作图检测到的SNP之间未发现共定位。
本研究首次比较了欧洲黑松中LA和LD作图方法,突出了这两种方法在解析育种计划中两个重要性状遗传结构方面的互补性。