Department of Biological Sciences, University of Pittsburgh, Pittsburgh, PA 15260, USA.
BMC Genomics. 2011 Jul 25;12:374. doi: 10.1186/1471-2164-12-374.
Statistics measuring codon selection seek to compare genes by their sensitivity to selection for translational efficiency, but existing statistics lack a model for testing the significance of differences between genes. Here, we introduce a new statistic for measuring codon selection, the Adaptive Codon Enrichment (ACE).
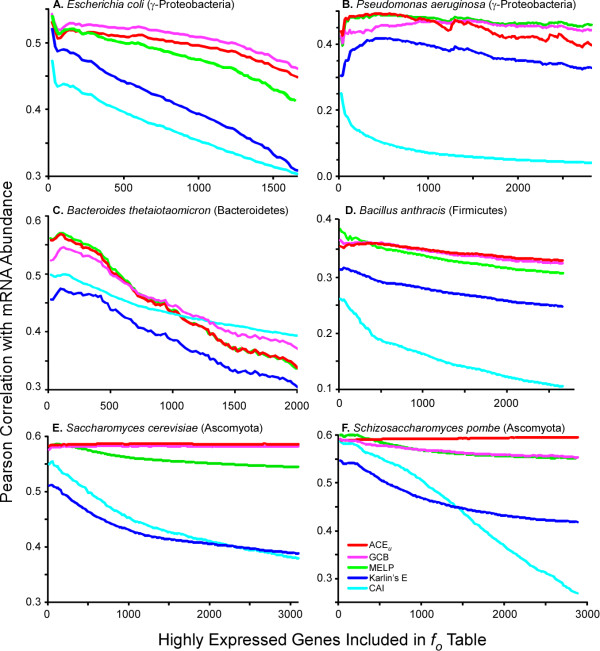
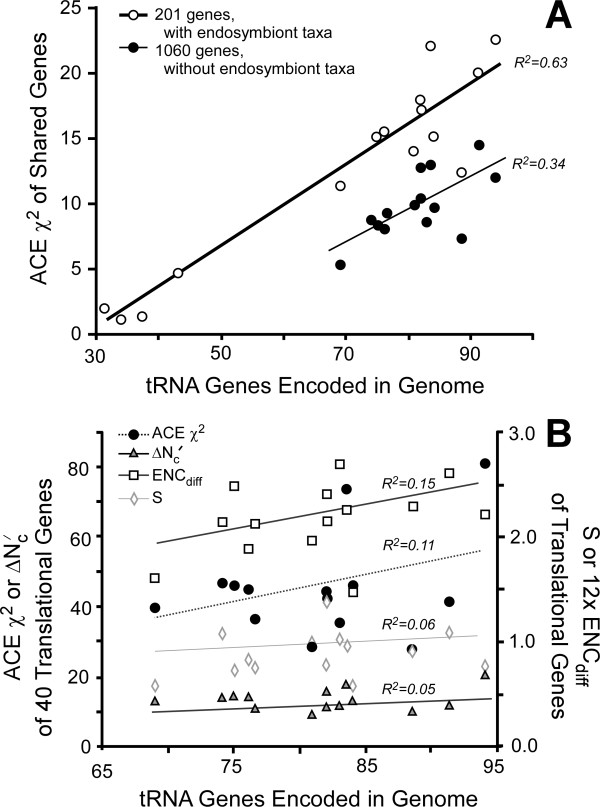
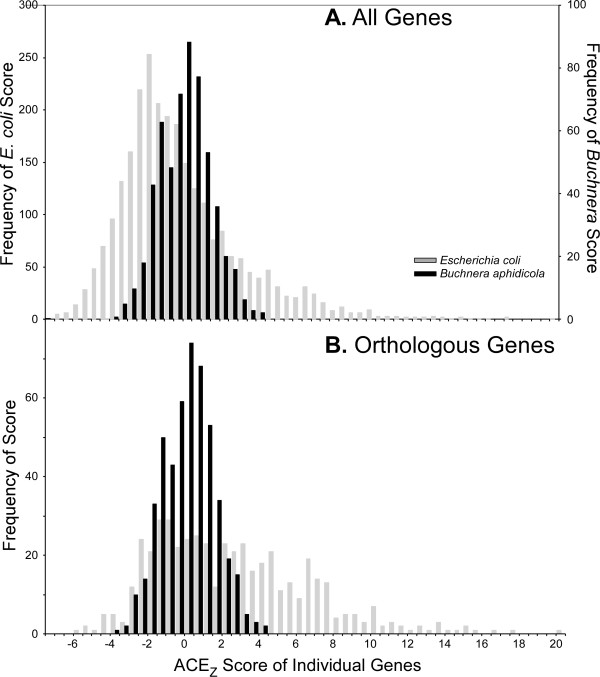
This statistic represents codon usage bias in terms of a probabilistic distribution, quantifying the extent that preferred codons are over-represented in the gene of interest relative to the mean and variance that would result from stochastic sampling of codons. Expected codon frequencies are derived from the observed codon usage frequencies of a broad set of genes, such that they are likely to reflect nonselective, genome wide influences on codon usage (e.g. mutational biases). The relative adaptiveness of synonymous codons is deduced from the frequency of codon usage in a pre-selected set of genes relative to the expected frequency. The ACE can predict both transcript abundance during rapid growth and the rate of synonymous substitutions, with accuracy comparable to or greater than existing metrics. We further examine how the composition of reference gene sets affects the accuracy of the statistic, and suggest methods for selecting appropriate reference sets for any genome, including bacteriophages. Finally, we demonstrate that the ACE may naturally be extended to quantify the genome-wide influence of codon selection in a manner that is sensitive to a large fraction of codons in the genome. This reveals substantial variation among genomes, correlated with the tRNA gene number, even among groups of bacteria where previously proposed whole-genome measures show little variation.
The statistical framework of the ACE allows rigorous comparison of the level of codon selection acting on genes, both within a genome and between genomes.
衡量密码子选择的统计数据旨在通过比较基因对翻译效率选择的敏感性来比较基因,但现有的统计数据缺乏测试基因之间差异显著性的模型。在这里,我们引入了一种新的衡量密码子选择的统计量,即适应性密码子富集(ACE)。
这个统计量以概率分布的形式表示密码子使用偏性,量化了相对于随机抽样的密码子的均值和方差,感兴趣基因中的偏好密码子过度出现的程度。预期的密码子频率是从广泛的基因集的观察到的密码子使用频率中得出的,使得它们可能反映出对密码子使用的非选择性、全基因组影响(例如突变偏性)。同义密码子的相对适应性是从相对于预期频率的预先选择的基因集中的密码子使用频率推断出来的。ACE 可以预测快速生长过程中的转录物丰度和同义替换率,其准确性可与现有指标相媲美或更高。我们进一步研究了参考基因集的组成如何影响统计数据的准确性,并建议了为任何基因组选择合适参考集的方法,包括噬菌体。最后,我们证明 ACE 可以自然地扩展,以定量衡量基因组范围内密码子选择的影响,这种方法对基因组中的很大一部分密码子都很敏感。这揭示了基因组之间存在显著差异,与 tRNA 基因数量相关,即使在以前提出的全基因组指标显示出很少变化的细菌群体中也是如此。
ACE 的统计框架允许严格比较基因组内和基因组间基因上密码子选择的水平。