Program in Cellular and Molecular Biology, University of Wisconsin - Madison, Madison, Wisconsin, United States of America.
PLoS One. 2011;6(7):e21614. doi: 10.1371/journal.pone.0021614. Epub 2011 Jul 18.
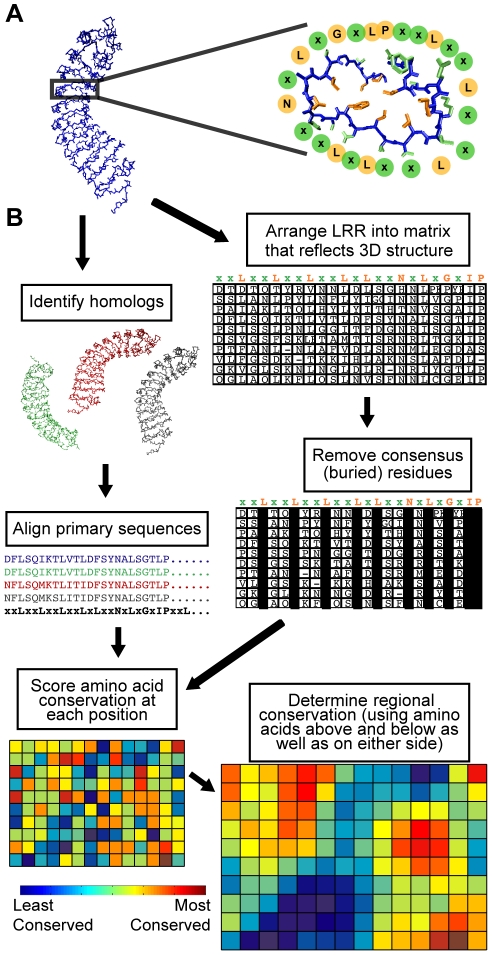
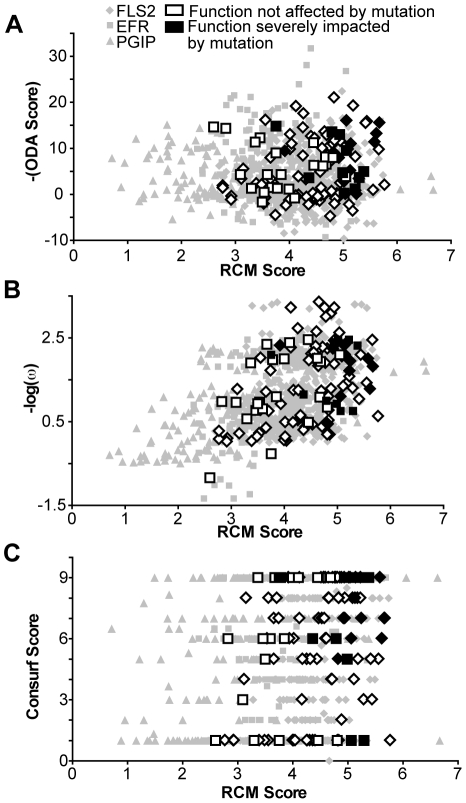
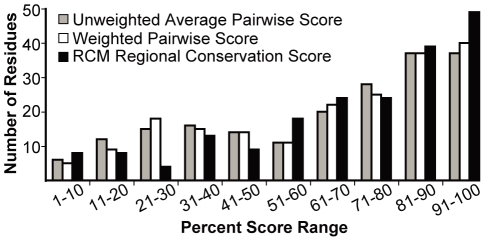
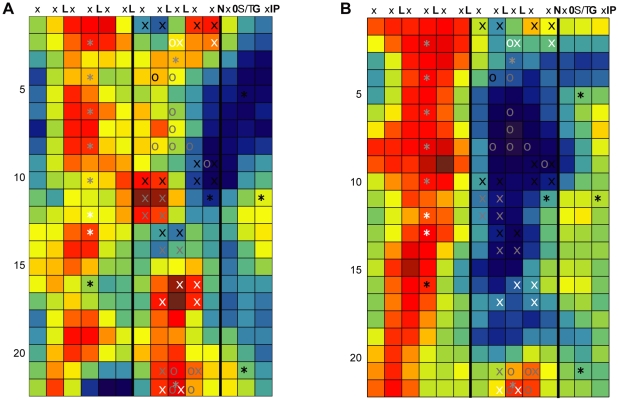
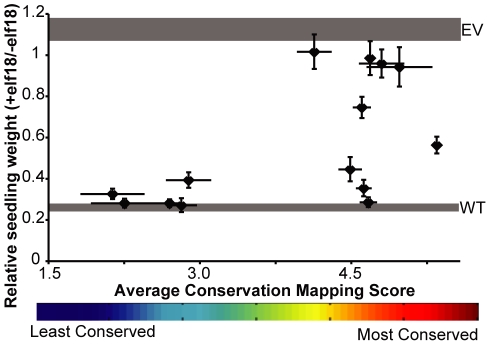
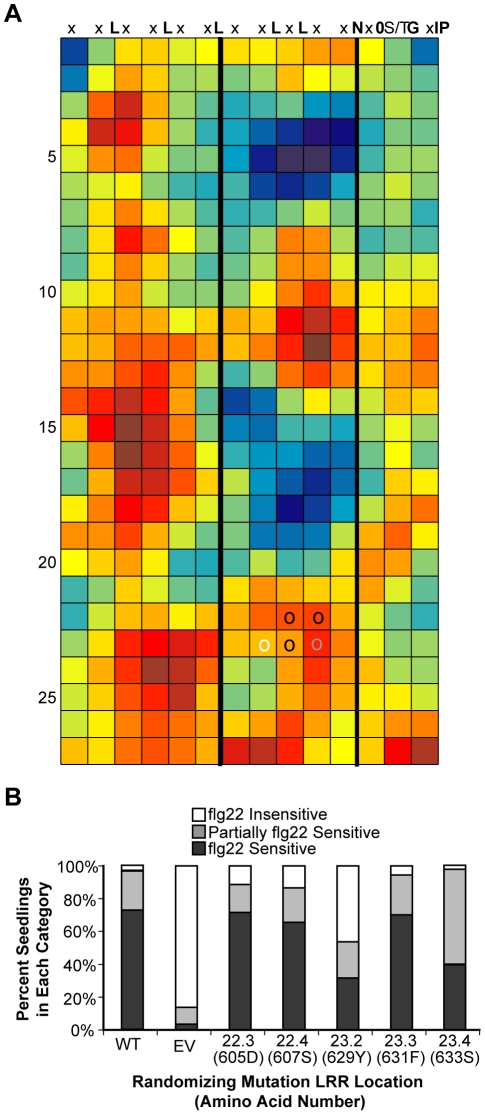
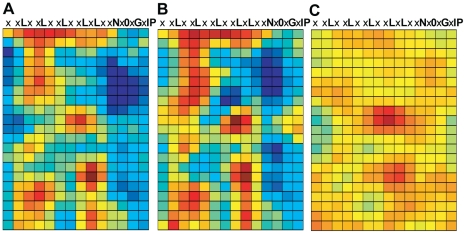
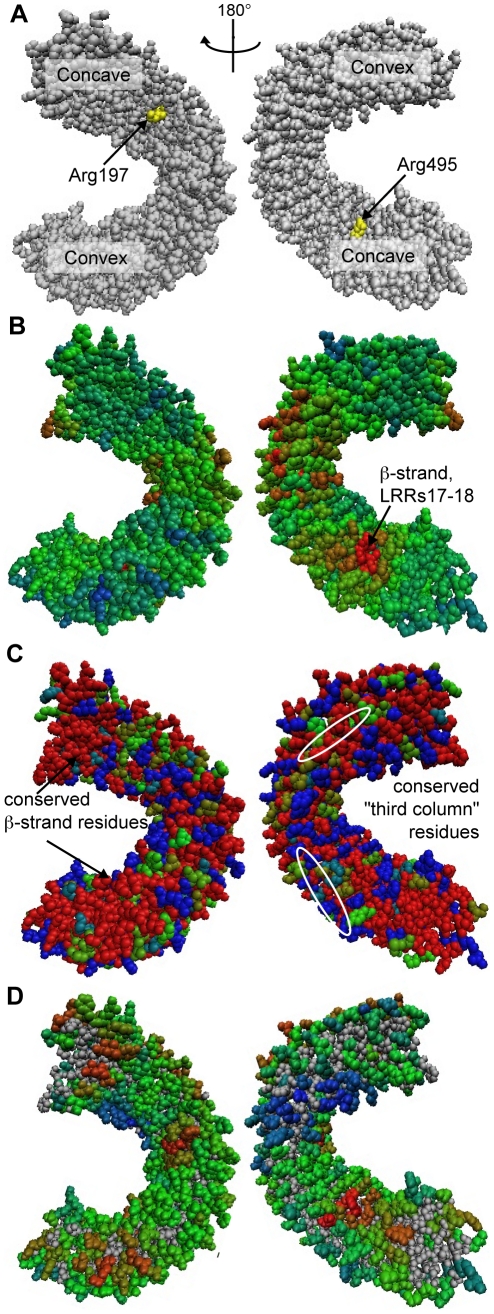
Computational prediction of protein functional sites can be a critical first step for analysis of large or complex proteins. Contemporary methods often require several homologous sequences and/or a known protein structure, but these resources are not available for many proteins. Leucine-rich repeats (LRRs) are ligand interaction domains found in numerous proteins across all taxonomic kingdoms, including immune system receptors in plants and animals. We devised Repeat Conservation Mapping (RCM), a computational method that predicts functional sites of LRR domains. RCM utilizes two or more homologous sequences and a generic representation of the LRR structure to identify conserved or diversified patches of amino acids on the predicted surface of the LRR. RCM was validated using solved LRR+ligand structures from multiple taxa, identifying ligand interaction sites. RCM was then used for de novo dissection of two plant microbe-associated molecular pattern (MAMP) receptors, EF-TU RECEPTOR (EFR) and FLAGELLIN-SENSING 2 (FLS2). In vivo testing of Arabidopsis thaliana EFR and FLS2 receptors mutagenized at sites identified by RCM demonstrated previously unknown functional sites. The RCM predictions for EFR, FLS2 and a third plant LRR protein, PGIP, compared favorably to predictions from ODA (optimal docking area), Consurf, and PAML (positive selection) analyses, but RCM also made valid functional site predictions not available from these other bioinformatic approaches. RCM analyses can be conducted with any LRR-containing proteins at www.plantpath.wisc.edu/RCM, and the approach should be modifiable for use with other types of repeat protein domains.
蛋白质功能位点的计算预测可以作为分析大型或复杂蛋白质的关键第一步。当代方法通常需要几个同源序列和/或已知的蛋白质结构,但许多蛋白质都没有这些资源。富含亮氨酸重复序列(LRR)是存在于所有分类群的许多蛋白质中的配体相互作用结构域,包括动植物的免疫系统受体。我们设计了重复保守性映射(RCM),这是一种预测 LRR 结构域功能位点的计算方法。RCM 利用两个或更多的同源序列和 LRR 结构的通用表示来识别预测 LRR 表面上保守或多样化的氨基酸补丁。RCM 使用来自多个分类群的已解决的 LRR+配体结构进行了验证,确定了配体相互作用位点。然后,RCM 用于从头剖析两种植物微生物相关分子模式(MAMP)受体,EF-TU 受体(EFR)和鞭毛素感应 2(FLS2)。通过 RCM 鉴定的位点对拟南芥 EFR 和 FLS2 受体进行的体内测试表明,存在以前未知的功能位点。RCM 对 EFR、FLS2 和第三种植物 LRR 蛋白 PGIP 的预测与 ODA(最佳对接区)、Consurf 和 PAML(正选择)分析的预测相当,但 RCM 还做出了其他生物信息学方法无法提供的有效功能位点预测。任何含有 LRR 的蛋白质都可以在 www.plantpath.wisc.edu/RCM 上进行 RCM 分析,并且该方法应该可以修改用于其他类型的重复蛋白结构域。