Cerebral Vascular Disease Research Center, Department of Neurology, Leonard M. Miller School of Medicine, University of Miami, Miami, Florida, United States of America.
PLoS One. 2011;6(7):e22057. doi: 10.1371/journal.pone.0022057. Epub 2011 Jul 15.
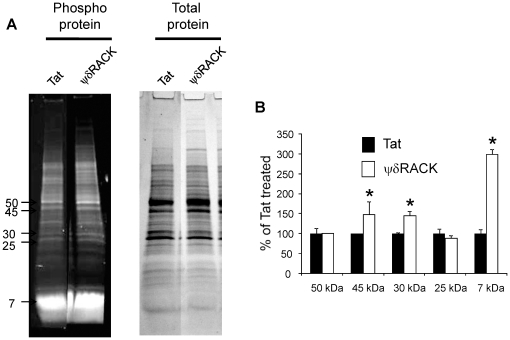
The release of cytochrome c from the mitochondria following cerebral ischemia is a key event leading to cell death. The goal of the present study was to determine the mechanisms involved in post-ischemic activation of protein kinase c delta (δPKC) that lead to cytochrome c release.
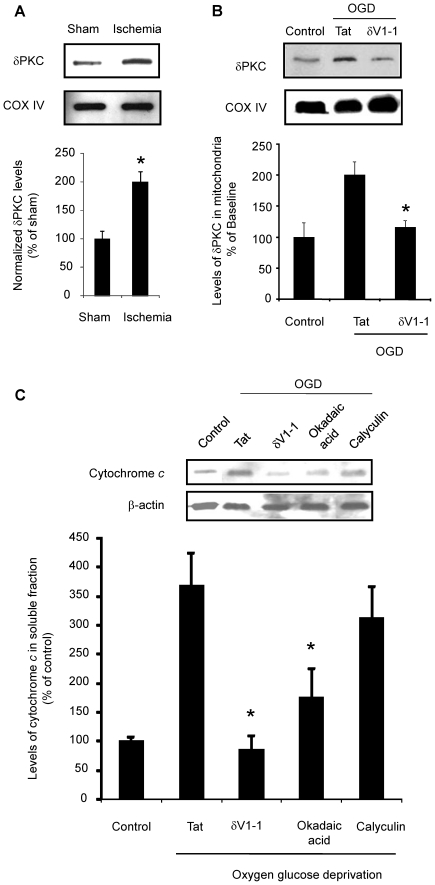
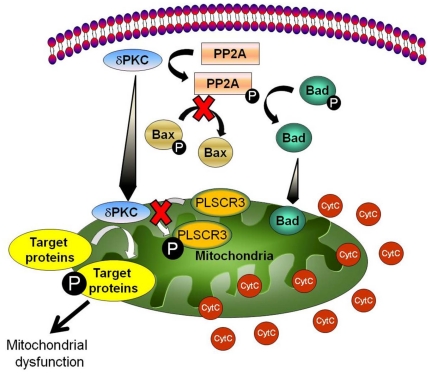
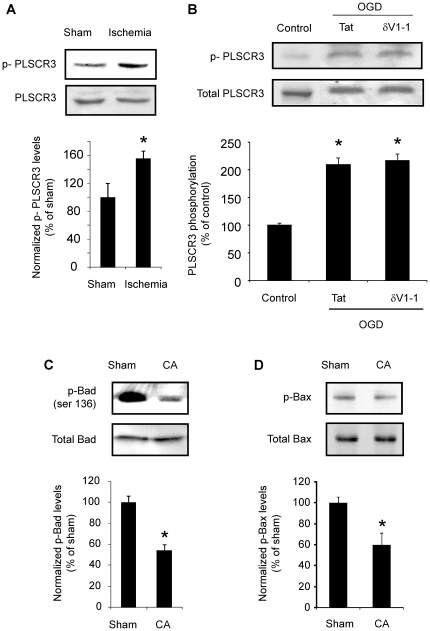
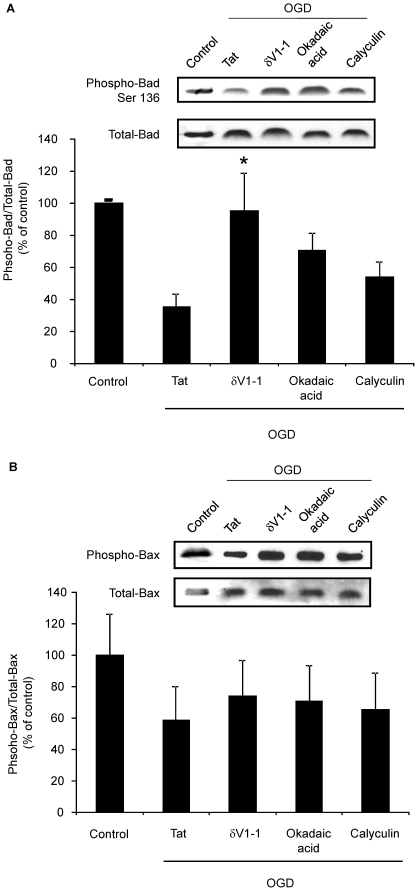
METHODS/FINDINGS: We used a rat model of cardiac arrest as an in vivo model, and an in vitro analog, oxygen glucose deprivation (OGD) in rat hippocampal synaptosomes. Cardiac arrest triggered translocation of δPKC to the mitochondrial fraction at 1 h reperfusion. In synaptosomes, the peptide inhibitor of δPKC blocked OGD-induced translocation to the mitochondria. We tested two potential pathways by which δPKC activation could lead to cytochrome c release: phosphorylation of phospholipid scramblase-3 (PLSCR3) and/or protein phosphatase 2A (PP2A). Cardiac arrest increased levels of phosphorlyated PLSCR3; however, inhibition of δPKC translocation failed to affect the OGD-induced increase in PLSCR3 in synaptosomal mitochondria suggesting the post-ischemic phosphorylation of PLSCR3 is not mediated by δPKC. Inhibition of either δPKC or PP2A decreased cytochrome c release from synaptosomal mitochondria. Cardiac arrest results in the dephosphorylation of Bad and Bax, both downstream targets of PP2A promoting apoptosis. Inhibition of δPKC or PP2A prevented OGD-induced Bad, but not Bax, dephosphorylation. To complement these studies, we used proteomics to identify novel mitochondrial substrates of δPKC.
We conclude that δPKC initiates cytochrome c release via phosphorylation of PP2A and subsequent dephosphorylation of Bad and identified δPKC, PP2A and additional mitochondrial proteins as potential therapeutic targets for ischemic neuroprotection.
脑缺血后细胞色素 c 从线粒体释放是导致细胞死亡的关键事件。本研究的目的是确定与缺血后蛋白激酶 c δ(δPKC)激活相关的机制,这些机制导致细胞色素 c 的释放。
方法/发现:我们使用心脏骤停的大鼠模型作为体内模型,以及体外类似物——氧葡萄糖剥夺(OGD)在大鼠海马突触体中。心脏骤停在再灌注 1 小时时触发 δPKC 向线粒体部分的易位。在突触体中,δPKC 的肽抑制剂阻断了 OGD 诱导的向线粒体的易位。我们测试了两种可能的途径,即 δPKC 激活如何导致细胞色素 c 的释放:磷脂 scramblase-3(PLSCR3)和/或蛋白磷酸酶 2A(PP2A)的磷酸化。心脏骤停增加了磷酸化 PLSCR3 的水平;然而,抑制 δPKC 易位未能影响突触体线粒体中 OGD 诱导的 PLSCR3 增加,表明缺血后 PLSCR3 的磷酸化不是由 δPKC 介导的。抑制 δPKC 或 PP2A 均可减少突触体线粒体中细胞色素 c 的释放。心脏骤停导致 Bad 和 Bax 的去磷酸化,Bad 和 Bax 都是 PP2A 的下游靶标,促进细胞凋亡。抑制 δPKC 或 PP2A 可防止 OGD 诱导的 Bad 去磷酸化,但不能防止 Bax 去磷酸化。为了补充这些研究,我们使用蛋白质组学鉴定了 δPKC 的新线粒体底物。
我们的结论是,δPKC 通过磷酸化 PP2A 启动细胞色素 c 的释放,随后导致 Bad 的去磷酸化,并确定 δPKC、PP2A 和其他线粒体蛋白作为缺血性神经保护的潜在治疗靶点。