Jantzen Stuart G, Sutherland Ben Jg, Minkley David R, Koop Ben F
Department of Biology & Centre for Biomedical Research, University of Victoria, Victoria, British Columbia, V8W 3N5, Canada.
BMC Res Notes. 2011 Jul 28;4:267. doi: 10.1186/1756-0500-4-267.
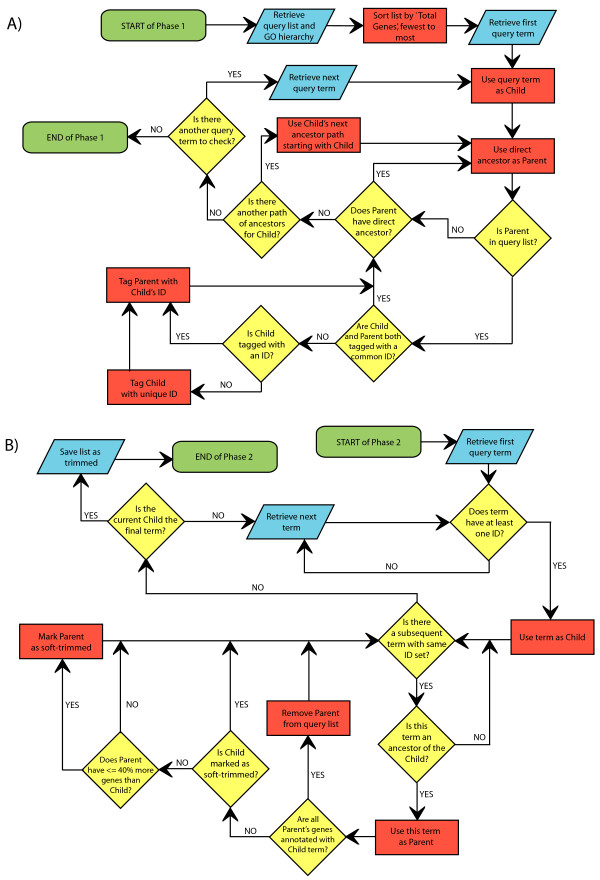
The increased accessibility of gene expression tools has enabled a wide variety of experiments utilizing transcriptomic analyses. As these tools increase in prevalence, the need for improved standardization in processing and presentation of data increases, as does the need to guard against interpretation bias. Gene Ontology (GO) analysis is a powerful method of interpreting and summarizing biological functions. However, while there are many tools available to investigate GO enrichment, there remains a need for methods that directly remove redundant terms from enriched GO lists that often provide little, if any, additional information.
Here we present a simple yet novel method called GO Trimming that utilizes an algorithm designed to reduce redundancy in lists of enriched GO categories. Depending on the needs of the user, this method can be performed with variable stringency. In the example presented here, an initial list of 90 terms was reduced to 54, eliminating 36 largely redundant terms. We also compare this method to existing methods and find that GO Trimming, while simple, performs well to eliminate redundant terms in a large dataset throughout the depth of the GO hierarchy.
The GO Trimming method provides an alternative to other procedures, some of which involve removing large numbers of terms prior to enrichment analysis. This method should free up the researcher from analyzing overly large, redundant lists, and instead enable the concise presentation of manageable, informative GO lists. The implementation of this tool is freely available at: http://lucy.ceh.uvic.ca/go_trimming/cbr_go_trimming.py.
基因表达工具的可及性提高,使得利用转录组分析进行的各种实验成为可能。随着这些工具的普及,对数据处理和呈现方面改进标准化的需求增加,防范解释偏差的需求也同样增加。基因本体论(GO)分析是解释和总结生物学功能的有力方法。然而,虽然有许多工具可用于研究GO富集,但仍需要直接从富集的GO列表中去除冗余术语的方法,这些冗余术语往往几乎不提供(如果有的话)额外信息。
在此,我们提出一种简单而新颖的方法,称为GO修剪,它利用一种算法来减少富集的GO类别列表中的冗余。根据用户需求,该方法可以不同的严格程度执行。在本文给出的示例中,最初的90个术语列表减少到了54个,消除了36个基本冗余的术语。我们还将此方法与现有方法进行比较,发现GO修剪虽然简单,但在整个GO层次结构深度的大数据集中,在消除冗余术语方面表现良好。
GO修剪方法为其他程序提供了一种替代方案,其中一些程序涉及在富集分析之前去除大量术语。该方法应使研究人员无需分析过于庞大、冗余的列表,而是能够简洁地呈现可管理、信息丰富的GO列表。此工具的实现可在以下网址免费获取:http://lucy.ceh.uvic.ca/go_trimming/cbr_go_trimming.py。