Department of Medicine, David Geffen School of Medicine, University of California, Los Angeles, CA, USA.
Cancer Res. 2011 Aug 1;71(15):5067-74. doi: 10.1158/0008-5472.CAN-11-0140.
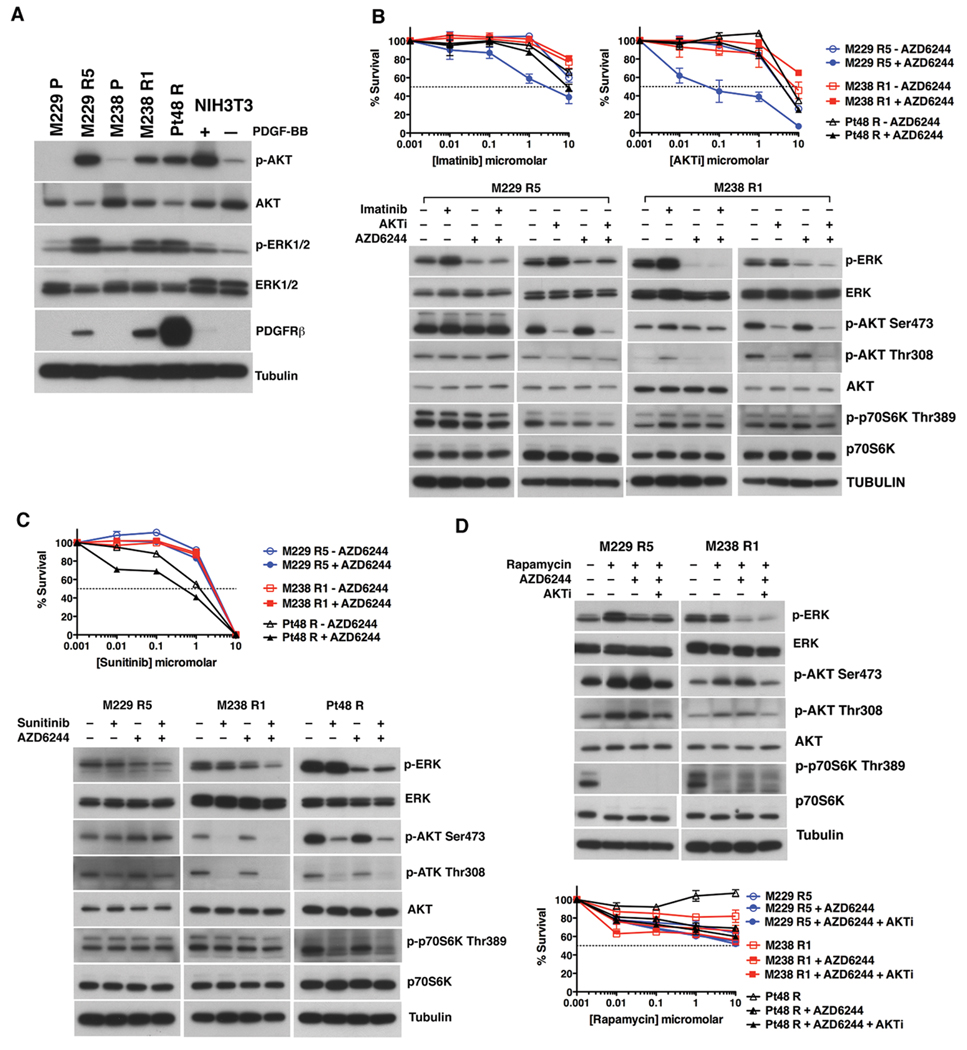
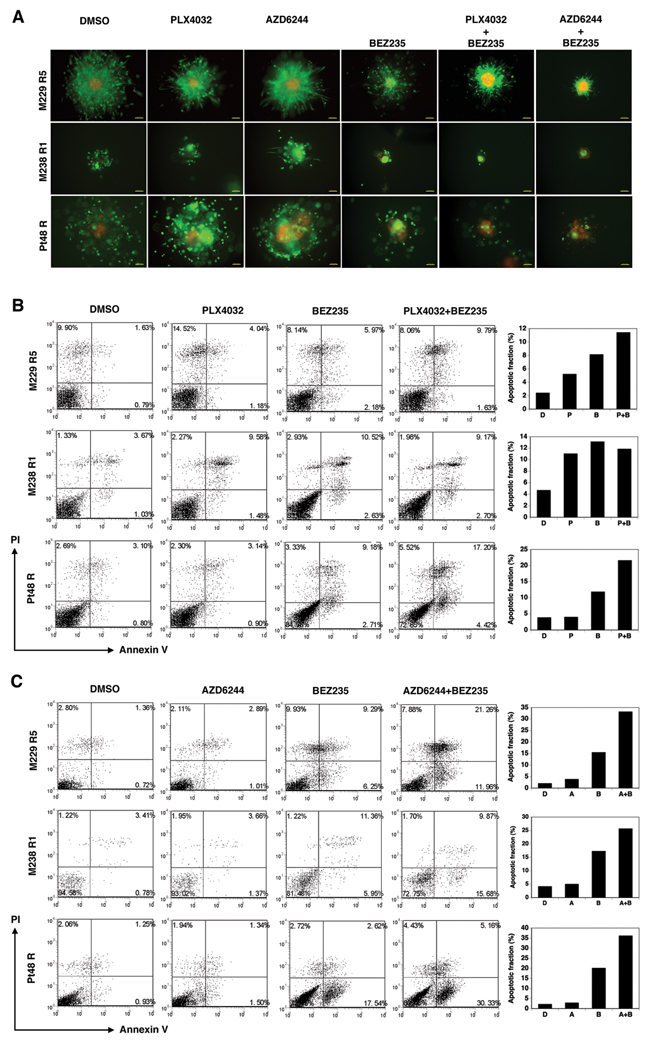
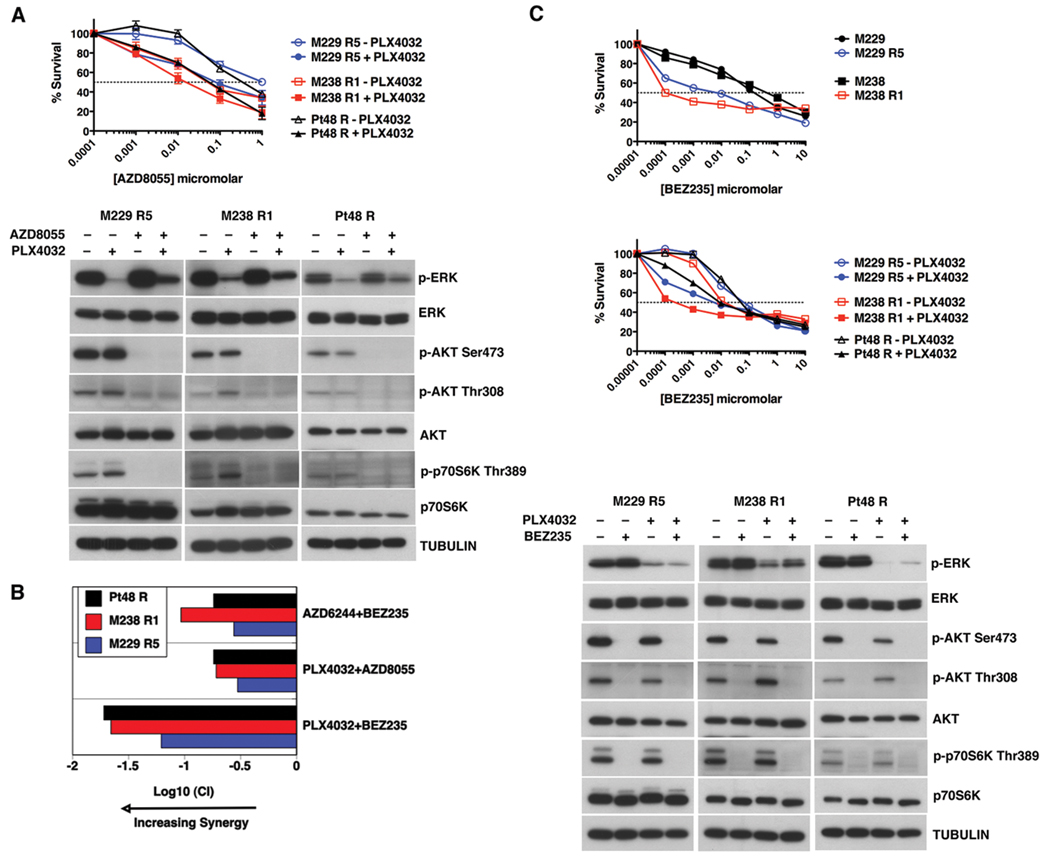
(V600E)B-RAF mutation is found in 50% to 60% of melanomas, and the novel agents PLX4032/vemurafenib and GSK2118436 that inhibit the (V600E)B-RAF kinase achieve a remarkable clinical response rate. However, as might be expected, acquired clinical resistance to these agents arises in most melanoma patients. PLX4032/vemurafenib resistance that arises in vivo in tumor matched short-term cultures or in vitro in melanoma cell lines is not caused by acquisition of secondary mutations in (V600E)B-RAF but rather is caused by upregulating platelet-derived growth factor receptor β (PDGFRβ) or N-RAS which results in resistance or sensitivity to mitogen-activated protein (MAP)/extracellular signal-regulated (ERK; MEK) kinase inhibitors, respectively. In this study, we define a targeted combinatorial strategy to overcome PLX4032/vemurafenib resistance in melanoma cell lines or short-term culture where the resistance is driven by PDGFRβ upregulation, achieving synergistic growth inhibition and cytotoxicity. PDGFRβ-upregulated, PLX4032-resistant (PPRM) cell lines show dual phospho (p)-ERK and p-AKT upregulation, and their growth inhibitory responses to specific small molecule inhibitors correlated with p-ERK, p-AKT, and p-p70S6K levels. Coordinate inhibition of (V600E)B-RAF inhibition and the RTK-PI3K-AKT-mTORC axis led to functionally significant rebound signaling, illustrating a robust and dynamic network connectivity. Combined B-RAF, phosphoinositide 3-kinase (PI3K), and mTORC1/2 inhibition suppressed both immediate early and delayed compensatory signaling, resulting in a highly synergistic growth inhibitory response but less efficient cytotoxic response. In contrast, the combination of MEK1/2, PI3K, and mTORC1/2 inhibitors consistently triggered apoptosis in a highly efficient manner. Together, our findings offer a rational strategy to guide clinical testing in preidentified subsets of patients who relapse during treatment with (V600E)B-RAF inhibitors.
(V600E)B-RAF 突变存在于 50%至 60%的黑色素瘤中,新型药物 PLX4032/vemurafenib 和 GSK2118436 可抑制 (V600E)B-RAF 激酶,获得显著的临床反应率。然而,正如预期的那样,大多数黑色素瘤患者会出现获得性临床耐药。在肿瘤匹配的短期培养物或黑色素瘤细胞系中体内出现的 PLX4032/vemurafenib 耐药性并不是由于 (V600E)B-RAF 获得继发性突变引起的,而是由于血小板衍生生长因子受体 β (PDGFRβ) 或 N-RAS 的上调引起的,这分别导致对丝裂原激活蛋白 (MAP)/细胞外信号调节激酶 (ERK; MEK) 激酶抑制剂的耐药或敏感。在这项研究中,我们定义了一种靶向联合策略,以克服黑色素瘤细胞系或短期培养物中由 PDGFRβ 上调驱动的 PLX4032/vemurafenib 耐药,实现协同生长抑制和细胞毒性。PDGFRβ 上调、PLX4032 耐药 (PPRM) 细胞系显示双重磷酸化 (p)-ERK 和 p-AKT 上调,其对特定小分子抑制剂的生长抑制反应与 p-ERK、p-AKT 和 p-p70S6K 水平相关。同时抑制 (V600E)B-RAF 抑制和 RTK-PI3K-AKT-mTORC 轴导致功能上显著的反弹信号,说明了强大而动态的网络连接。联合 B-RAF、磷酸肌醇 3-激酶 (PI3K) 和 mTORC1/2 抑制抑制了即时早期和延迟补偿信号,导致高度协同的生长抑制反应,但细胞毒性反应效率较低。相比之下,MEK1/2、PI3K 和 mTORC1/2 抑制剂的联合始终以高效的方式触发细胞凋亡。总之,我们的研究结果为指导针对治疗期间复发的 (V600E)B-RAF 抑制剂的预先确定患者亚组的临床测试提供了合理的策略。